2024/03/24 ローカルでのラン例追記
微生物は、二次代謝や特殊な代謝の一環として、小さな生物活性化合物を生成する。このような代謝物は、抗菌、抗がん、抗真菌、抗ウイルスなどの生物活性を持つことが多く、医療や農業への応用に重要な役割を担っている。過去10年間、ゲノムマイニングは、これらの化合物の利用可能な生物多様性を探索、アクセス、分析するために広く使用されている手法となっている。2011年以来、「antibiotics and secondary metabolite analysis shell-antiSMASH」(https://antismash.secondarymetabolites.org/)は、OSI承認のオープンソースライセンスのもと、無料で使用できるウェブサーバーおよびスタンドアロンツールとして、研究者の微生物ゲノムマイニング作業をサポートしてきた。現在、古細菌、細菌、真菌における生合成遺伝子クラスター(BGC)の検出と特徴付けに最も広く使われているツールである。AntiSMASH 7は、サポートするクラスターの種類を71から81に増やし、化学構造予測、酵素アセンブリラインの可視化、遺伝子クラスター制御の分野での改良を含んでいる。
Documentation
https://docs.antismash.secondarymetabolites.org/
v.7の新機能及びバグ修正
https://docs.antismash.secondarymetabolites.org/changelog/7.0/
bacteria
https://antismash.secondarymetabolites.org/#!/start
Fungi
https://fungismash.secondarymetabolites.org/#!/start
将来的なantiSMASHの機能を試すためのウェブサイトの提供も開始された
https://experimentalsmash.secondarymetabolites.org/
ここではbacteriaの
https://antismash.secondarymetabolites.org/#!/startにアクセスする。

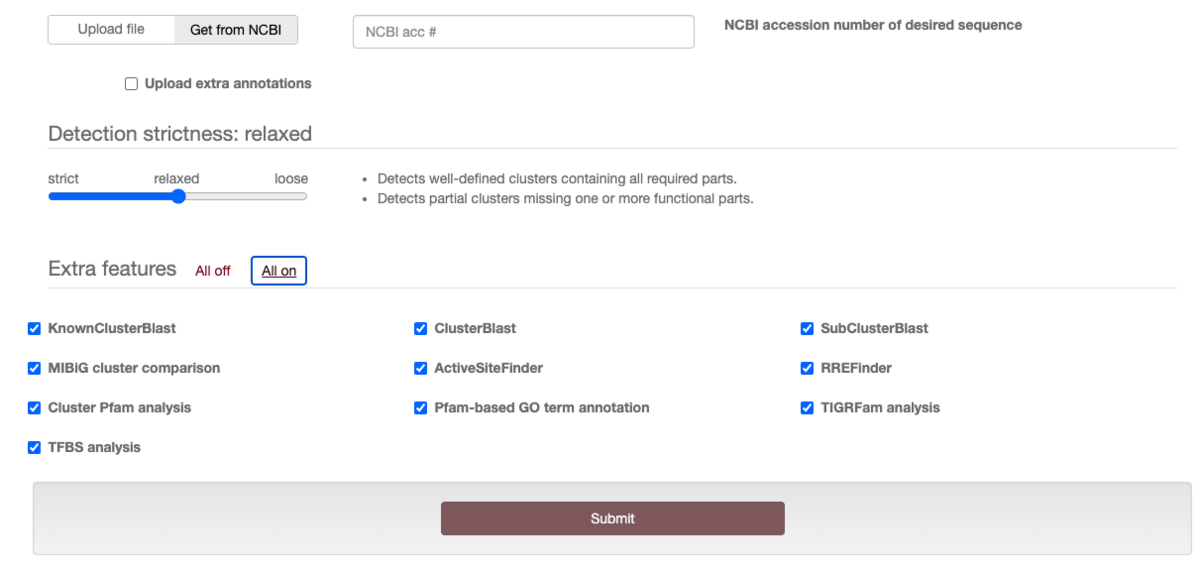
Biosynthetic Gene Clusters (BGCs)を予測するには、遺伝子クラスターを調べたいゲノムのGenbankファイル、GFF3ファイルをアップロードするか、NCBI accession numberを指定する。
追加の解析を行うオプションも用意されている。All onで全てONになる。
exampleの結果を見てみる。
https://antismash.secondarymetabolites.org/upload/example/index.html#
予測された二次代謝産物クラスターが表示される。右端にはsimilarity (%)も表示されている。
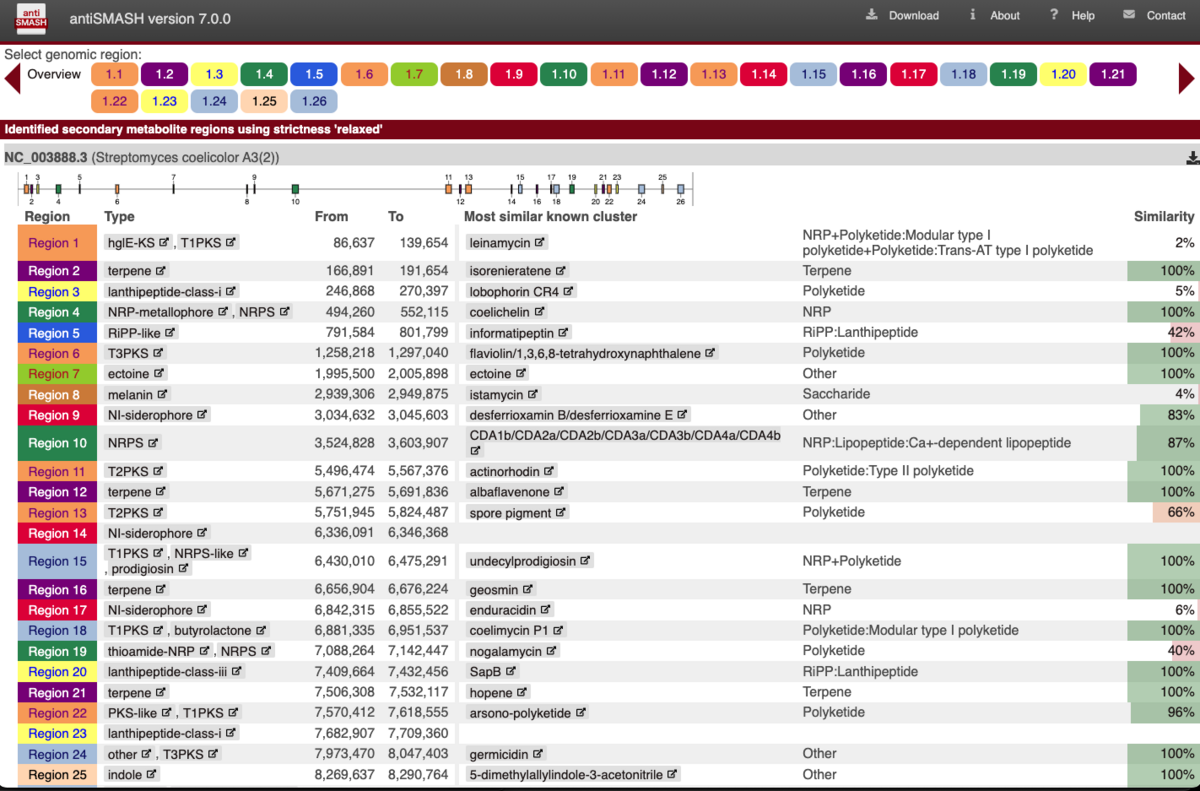
クリックすると詳細が表示される。

画面中央のタブから追加解析の結果に切り替えることができるようになっている。いくつか見てみる。
Gene overview
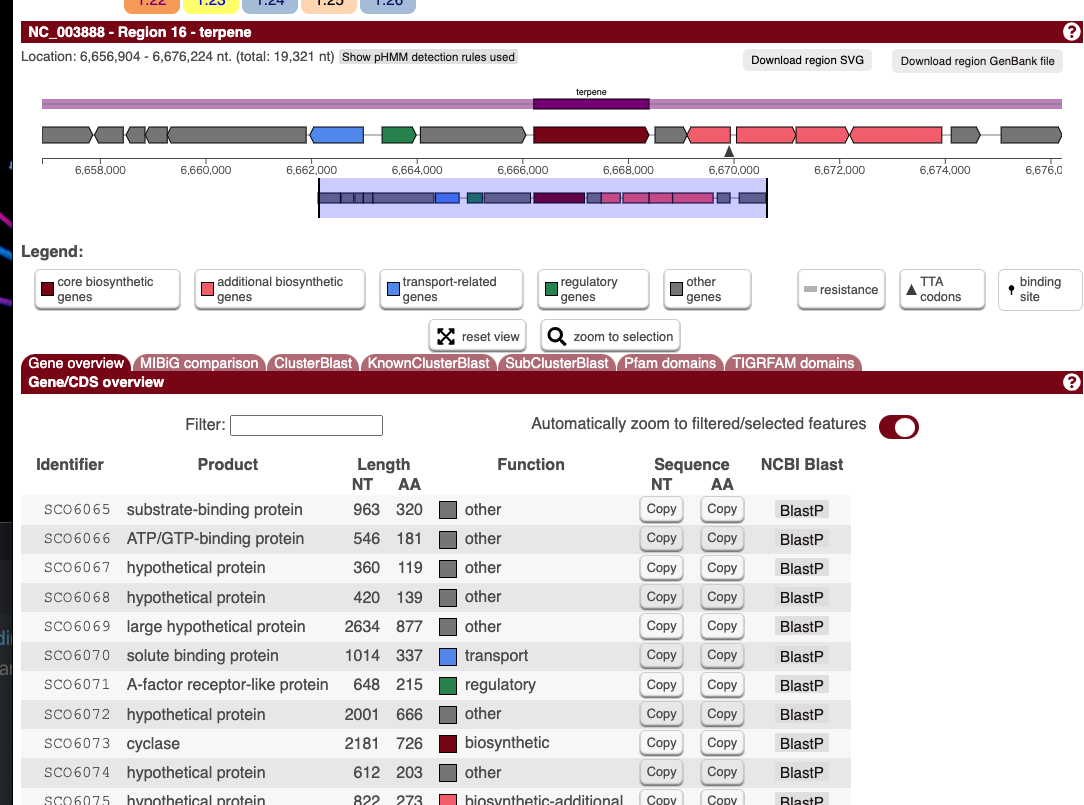
クラスターを構成する遺伝子の配列をコピーしたり、BlastPサーチできるようになっている。
nrps_pks_domains
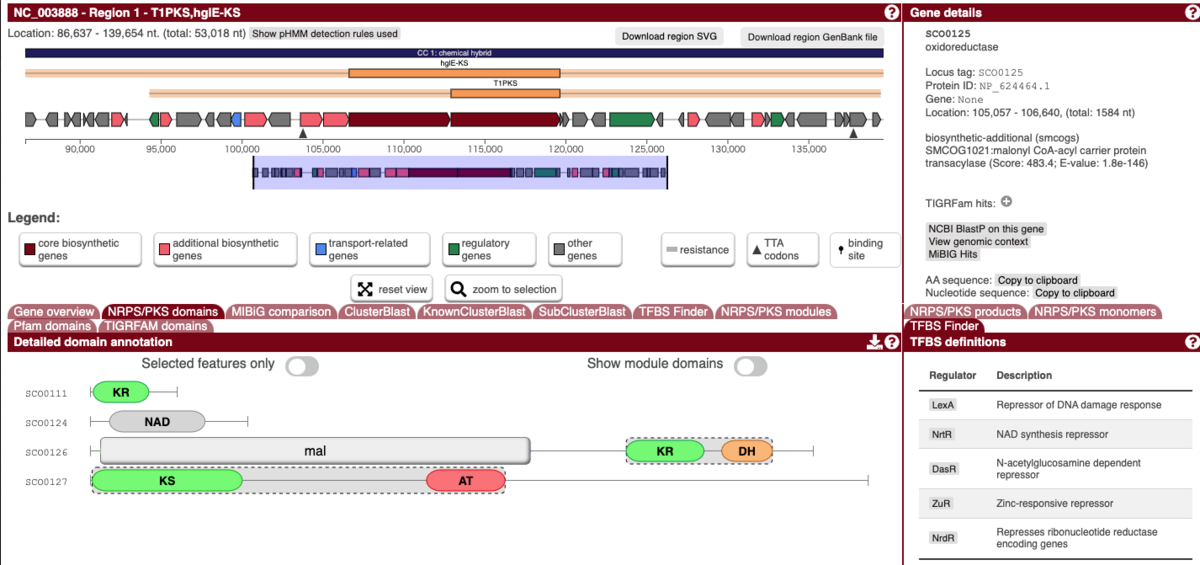
pfam domain

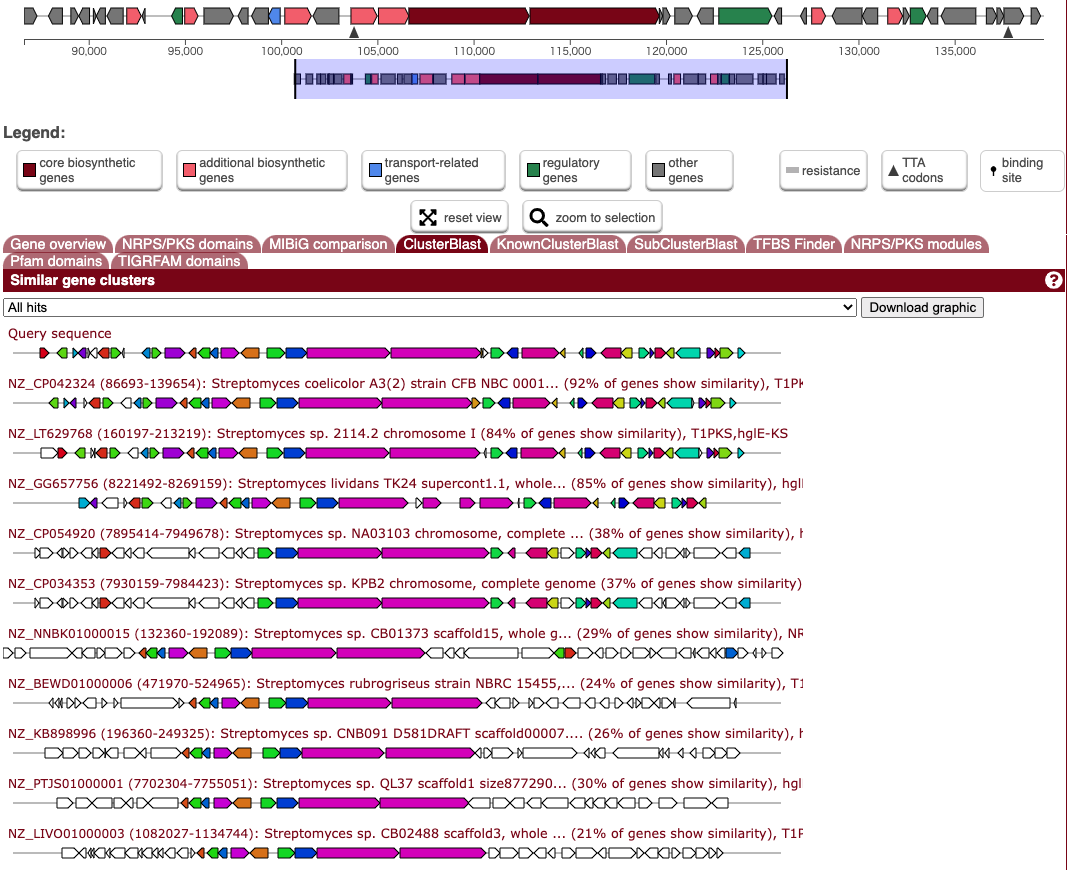
ClusterBlast;他のゲノムでの類似したクラスターの探索
TFBS Finder;v.7の新機能の1つ。LogoMotifプロファイルを使用して転写因子結合部位を見つける。

詳細;https://docs.antismash.secondarymetabolites.org/modules/tfbs/
ローカルマシンへのインストール
これまでのバージョン同様、ローカルのCLI環境では詳細なオプションを設定してランできる。大規模な解析にも向いている。
#bioconda (link)
#依存が多いので環境を作って導入するのが無難
mamba create -n antismash -y
conda activate antismash
mamba install -c bioconda antismash -y
#database
download-antismash-databases
> antismash
$ antismash -h
########### antiSMASH 7.1.0 #############
usage: antismash [-h] [options ..] sequence
arguments:
SEQUENCE GenBank/EMBL/FASTA file(s) containing DNA.
--------
Options
--------
Help options:
-h, --help Show basic help text.
--help-showall Show full list of arguments.
Basic analysis options:
-t {bacteria,fungi}, --taxon {bacteria,fungi}
Taxonomic classification of input sequence. (default: bacteria)
-c CPUS, --cpus CPUS How many CPUs to use in parallel. (default for this machine: 128)
--databases PATH Root directory of the databases (default: /home/kazu/mambaforge/envs/antismash/lib/python3.9/site-packages/antismash/databases).
Output options:
--output-dir OUTPUT_DIR
Directory to write results to.
--output-basename OUTPUT_BASENAME
Base filename to use for output files within the output directory.
--html-title HTML_TITLE
Custom title for the HTML output page (default is input filename).
--html-description HTML_DESCRIPTION
Custom description to add to the output.
--html-start-compact Use compact view by default for overview page.
--html-ncbi-context, --no-html-ncbi-context
Show NCBI genomic context links for genes (default: False). (default: False)
Additional analysis:
--fullhmmer Run a whole-genome HMMer analysis using Pfam profiles.
--cassis Motif based prediction of SM gene cluster regions.
--clusterhmmer Run a cluster-limited HMMer analysis using Pfam profiles.
--tigrfam Annotate clusters using TIGRFam profiles.
--asf Run active site finder analysis.
--cc-mibig Run a comparison against the MIBiG dataset
--cb-general Compare identified clusters against a database of antiSMASH-predicted clusters.
--cb-subclusters Compare identified clusters against known subclusters responsible for synthesising precursors.
--cb-knownclusters Compare identified clusters against known gene clusters from the MIBiG database.
--pfam2go Run Pfam to Gene Ontology mapping module.
--rre Run RREFinder precision mode on all RiPP gene clusters.
--smcog-trees Generate phylogenetic trees of sec. met. cluster orthologous groups.
--tfbs Run TFBS finder on all gene clusters.
Lowest GC content to annotate TTA codons at (default: 0.65).
Gene finding options (ignored when ORFs are annotated):
--genefinding-tool {glimmerhmm,prodigal,prodigal-m,none,error}
Specify algorithm used for gene finding: GlimmerHMM, Prodigal, Prodigal Metagenomic/Anonymous mode, or none. The 'error' option will raise an error if genefinding is attempted. The 'none' option will not run genefinding. (default: error).
--genefinding-gff3 GFF3_FILE
Specify GFF3 file to extract features from.
#GenBankを指定
antismash -c 20 --output-dir outdir input.gbk
#fastaを指定。prodigalで遺伝子予測する。20スレッド指定
antismash -c 20 --genefinding-tool prodigal --output-dir outdir input.fasta
#GFF3を指定、真菌、20スレッド指定
antismash -c 20 -t fungi --genefinding-gff3 --output-dir outdir input.gff3
- -c How many CPUs to use in parallel. (default for this machine: 128)
-
-t {bacteria, fungi} Taxonomic classification of input sequence. (default: bacteria)
-
--genefinding-tool {glimmerhmm, prodigal, prodigal-m, none, error}
Specify algorithm used for gene finding: GlimmerHMM, Prodigal, Prodigal Metagenomic/Anonymous mode, or none. The 'error' option will raise an error if genefinding is attempted. The 'none' option will not run genefinding. (default: error).
-
--genefinding-gff3 Specify GFF3 file to extract features from.
こちらで説明されています。
https://docs.antismash.secondarymetabolites.org/install/
引用
antiSMASH 7.0: new and improved predictions for detection, regulation, chemical structures and visualisation
Kai Blin, Simon Shaw, Hannah E Augustijn, Zachary L Reitz, Friederike Biermann, Mohammad Alanjary, Artem Fetter, Barbara R Terlouw, William W Metcalf, Eric J N Helfrich, Gilles P van Wezel, Marnix H Medema, Tilmann Weber
Nucleic Acids Research, Published: 04 May 2023
関連