2022/01/30 誤字修正
主成分分析(PCA)は、RNAシーケンス(RNA-seq)遺伝子発現アッセイなどの高次元データにおける品質評価や探索的分析に、ゲノミクスアプリケーションで頻繁に使用されている。この目的のために開発された多くのソフトウェアパッケージがあるにもかかわらず、これらの操作を実行するための対話的で包括的なインタフェースは不足している。pcaExplorerはShinyフレームワークを用いてRで実装されており、オープンソースのBioconductorプロジェクトのデータ構造を利用している。ユーザーは、一般的な概要、サンプルや遺伝子の次元削減、主成分の機能的解釈など、利用可能なさまざまなモジュールで発現データを評価しながら、出版に適したさまざまなグラフを簡単に作成することができる。pcaExplorerは、Bioconductorプロジェクト(http://bioconductor.org/packages/pcaExplorer/)のRパッケージとして配布されており、インタラクティブなデータ探索の重要なステップにおいて、幅広い研究者を支援するように設計されている。
pcaExplorerは全てRプログラミング言語で書かれており、Bioconductorから入手可能な他のいくつかの広く使われているRパッケージに依存している。主な機能は、pcaExplorer()関数を1回呼び出すだけでアクセスでき、ウェブアプリケーションが起動する。pcaExplorerは、macOS、Linux、およびWindowsでテストされている。Bioconductor のプロジェクトページ(http://bioconductor.org/packages/pcaExplorer/)からダウンロードでき、その開発版は https://github.com/federicomarini/pcaExplorer/ に掲載されている。pcaExplorerを様々なデータセットに対して実行するには、少なくとも8GBのRAMを搭載した典型的な最新のラップトップまたはワークステーションで十分である。ロードと前処理のステップはデータセットのサイズによって異なるが、pcaExplorerのセッションを完了するのに必要な時間は、主に探索の深さに依存する。ユーザーがパッケージとそのインターフェースに慣れれば、典型的なセッションは約15~30分(レポート作成を含む)かかると予想される。
pcaExplorerはローカルにインストールするだけでなく、WebアプリケーションとしてShinyサーバーにデプロイすることもでき、ユーザーは追加のソフトウェアをインストールすることなく、データを探索することができる。典型的な例としては、バイオインフォマティシャンやITシステム管理者が、同じ研究グループのメンバー向けにインスタンスを提供したり、関心のあるデータセットの関連機能を探索・表示できるようにできる。サーバー上でpcaExplorerをセットアップする完全な手順を説明するために、GitHubリポジトリhttps://github.com/federicomarini/pcaExplorer_serveredition ですべてのステップを文書化している。ウェブサービスと比較して、本Shinyアプリ(およびサーバ)アプローチは、機密データアクセスを制御するために、機関のファイアウォール内で保護されたデプロイメントを可能にする。(一部略)将来的には、他の次元削減技術(例えば、スパースPCA [46]やt-SNE [47]など)も検討する予定である。
manual
https://federicomarini.github.io/pcaExplorer/
チュートリアル(↓でリンクしている公開インスタンスのデモデータが使われている)
Up and running with `r Biocpkg("pcaExplorer")` • pcaExplorer
一般に公開されているインスタンス
デモンストレーションの目的で、プライマリーヒト気道平滑筋細胞株データセット(論文の引用#39)にアクセスすることができる。
http://shiny.imbei.uni-mainz.de:3838/pcaExplorer/
インストール
condaコマンドで作成した仮想環境にrstudio free版と一緒に導入した(R3.6.1使用)。
#conda (bioconda)
mamba create -n pcaexplorer -y
conda activate pcaexplorer
mamba install -c bioconda bioconductor-pcaexplorer -y
mamba install -c r rstudio -y
#shiny GUIなどでデモデータも読み込むならairway(link)と-org.hs.eg.dbパッケージが必要
mamba install -c bioconda bioconductor-airway -y
mamba install -c bioconda bioconductor-org.hs.eg.db -y
#もしくはBiocManagerを使う(condaで利用可能なバージョンはやや古いことがある)。
if (!requireNamespace("BiocManager", quietly=TRUE))
install.packages("BiocManager")
BiocManager::install("pcaExplorer")
#dockerhubのイメージ(おそらく公式ではない、試してません)
https://hub.docker.com/r/chrischeshire/pcaexplorer/tags
> browseVignettes("pcaExplorer")

実行方法
$ rstudioとタイプしてrstudioのGUIを立ち上げる。
rstudioのRコンソールにて、pcaExplorerのパッケージをロードする。
library("pcaExplorer")
shiny のGUIを立ち上げる。入力データはGUIを立ち上げてから読み込んでも良いが、立ち上げる時に読み込むこともできる。
#データを指定せずに立ち上げる
pcaExplorer()
#HTSeq-countやfeatureCountsなどで得たリードカウントマトリックスを指定する(countmatrix)。coldata は条件、組織、細胞株、ランバッチなどの実験の共変量を含むデータフレーム。
pcaExplorer(countmatrix = countmatrix, coldata = coldata)
#DEseq2の解析結果を読み込む
pcaExplorer(dds = dds, dst = dst)
立ち上がった。
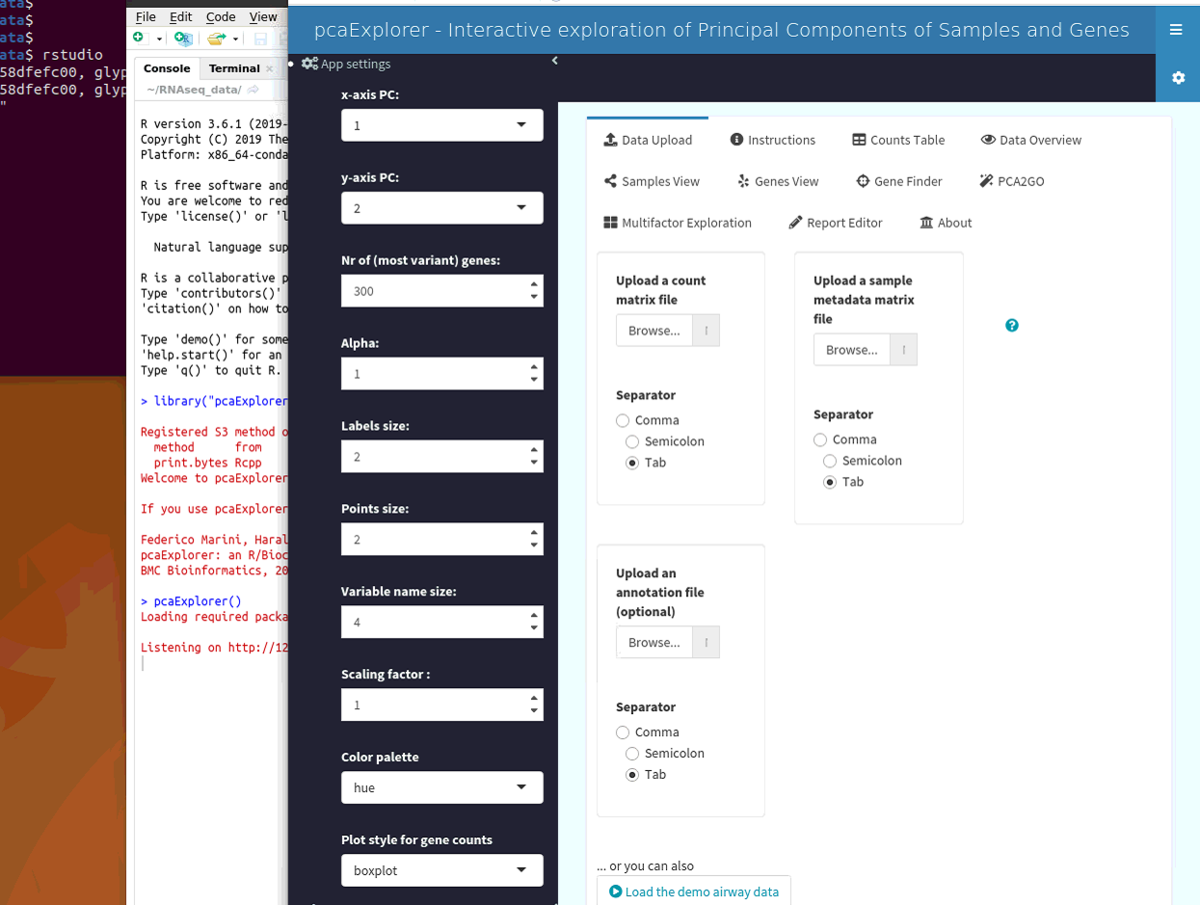
Data Uploadタブ
データを指定していない時は、Data Uploadタブでアップロードする。

pcaExplorerを使用するには、HTSeq-count や featureCounts などのツールでリードを遺伝子などで生成した生のカウント行列と、目的のサンプルの必須変数(条件、組織、細胞株、シーケンスラン、バッチ、ライブラリタイプなど)を含む実験メタデータテーブルの二つが必要となる。
カウントマトリックスとメタデータテーブルは、行名を識別子とし、サンプルのIDを示すヘッダーを付けたタブかカンマ、もしくはセミコロン区切りのテキストファイルとして提供する。
カウントデータの場合:特徴は行に、サンプルは列に格納される。

ヘッダーはddsとdstオブジェクトの構築とその有効性のチェックに使用されるため重要。例えば、countマトリックスの列名は、サンプルメタデータの行名と同一でなければならない。
メタデータの例
メタデータの各行には、各サンプルの関連する実験変数が格納される。

サンプル名はカウントデータと一致している必要がある。
annotationファイルの例
任意で提供する遺伝子アノテーションでは、各行は遺伝子)で、各行の1列目にはその ID、2列目には(gene_name )には読みやすい形式(例:HGNC 遺伝子記号)を含める。

annotationファイルは任意だが、提供すれば、遺伝子記号ではなくてより人が読みやすい遺伝子名を利用できるようになる。
アップロード後、画面のプレビューボタン(下の緑の四角部分)を押すとアップロードしたデータを確認できる。

メタデータも同様にプレビューできる。

データが用意できたら、最後にGenerate the dds and dst objectsボタン(右端の緑ボタン)をクリックする。ddsはDESeqDataSetオブジェクト。発現解析の入力値、中間計算、結果を格納するために使用される。dstはDESeqTransform オブジェクト。

それから、画面下の正規化(3つある青ボタン)のいずれかを実行する。デモデータでは、左端の分散安定化変換が選択される。

トランスクリプトームデータセットのサンプルによる平均-分散依存性を低減する目的で、異なるデータ変換を適用することができるようになっている。
ここではairway(link)のexample dataを使う。Load the demo~をクリック後、右下のall objects~が出れば準備完了となる。

Counts Tableタブ
このタブでは、transform(ここでは正規化)したデータをプレビューしたり、画面下のヒートマップでサンプル間の距離や相関を調べることができる。

Runボタンを押すとサンプル間散布図マトリックスとヒートマップが表示される。


Data Overviewタブ
サンプル間ヒートマップ。マンハッタン距離が選択されているが、ユークリッド距離や相関ベース距離も選択できる。




基本的なサマリー情報も表示される。”Threshold on the row sums of the counts or the row means of the normalized counts (more stringent) ”の数値を0から変更すると何個の遺伝子が検出されるか調べることができる。
Sample viewタブ
サンプルビューは、文字通りサンプル(ユーザー提供のメタデータ)に注目したPCAビューになる。左側のメニューで選択した因子に着目してPCAの結果を調べることができる。

このデータはPC1とPC2で80%を超えている。
左のメニューからcellに変更した。ラベルはそのままだが、色付けがcellの情報に代わり、判例もcellに切り替わっている。下の図から、PC2(y軸)はcellの種類がデータセット内の変動のかなりの部分を説明することが分かる。一方で、PC1はcellの種類では説明出来なさそうなことも分かる。

次にdex処理(treatedとuntreated)に切り替えた。PC1(x軸)の変動については、この処理の有無で説明できそうなことが分かる。

PC1とPC2だけでは寄与率が低い場合、より低い次元も同様にして調べていく。このように、pcaExplorerはサンプル間の変動の説明変数を視覚化して調べてために活躍する。
(多因子を考慮した複雑なRNA seq解析では、発現変動遺伝子を取り出す前に、主要な因子を明らかにしてモデルに取り込む必要がある。その確認にも役立つ。)
右の棒グラフは各PCの寄与率推移を示している。説明変数が他にもあるなら、PC4までは調べたほうが良さそうに見える。

draw a confidence~にチェックを付けた。

下のスクロールすると、各PCで負荷量が高いtop10とbottom10の遺伝子を確認できる。画面ではtop20に増やしている。上がPC1、下がPC2。

PCAは異常なサンプルを判別するためにも活躍する。上の分析で奇妙なサンプルプロットがみつかれば、それを外れ値とみなし、サンプルを一時的に除く機能もある。除くとPCAプロットが変化して、効果をその場で確認できる。

コメント;外れ値サンプルといっても色々な意味がある。サンプル調整、HTSシークエンシングに失敗しているのかもしれないが、それは、解析前に気づく可能性がある。より重要なのは、サンプルロットが質的に違った(*1)、他の組織が混じっていた、見えない汚染があったなどで、これらはデータを分析しない限りは分からないし、几帳面に実験を行っていても起こり得る。外れ値サンプルが残っていると反復内の分散が大きくなるので、クラシカルな2群間比較で発現変動遺伝子を調べてもほぼゼロになる。よって、PCAなどの方法で前もって異常なサンプルを分析するのは重要な意味を持つ。その時の状況によっては、発現変動遺伝子を取り出すのは危険と判断して(本質的に比較する組み合わせ間で発現プロファイルの差がないのかもしれない)、一部の比較は諦め、残ったサンプルの組み合わせだけで発現解析を進めなければいけないこともある。
一番下の3D plotビューでは、3つめの次元も追加して調べることができる。ここではPC1、PC2、PC3。

画面は自由に動かすことができる。
Genes Viewタブ
遺伝子ビューでは、先ほどの実験因子で大きく変動した遺伝子など、遺伝子に着目して調べることができる。
左のメニューで選択したtop300の変動の遺伝子のPCAプロット。発現パターンが類似してる遺伝子が近くにプロットされる。

サンプル(左のメニューから選択)は遺伝子バイプロット内の矢印として表示される。どの遺伝子が同様の挙動かを示す。
図をドラッグして囲むと(下図左上のグラフの水色の四角)右に拡大された図が表示され、囲まれた範囲内の遺伝子のサンプル間発現プロファイルが下に表示される。

説明変数dexにして、dexの矢印先の遺伝子を拡大した。dex条件で抑制されていることが分かる(下の水色サンプル)。

このように、遺伝子ビューでは、各PCで主要な変動をしている遺伝子を調べることができる。
GeneFinderタブ
重要な遺伝子が分かったら、GeneFinderでその遺伝子の発現量を調べることができる。調べるには遺伝子名を打ち込む。

box plotかviolin plotで表示される(左のメニューから切り替える)。ただし、アノテーションファイルで略称を提供していない場合はフルのgene IDを提供する必要がある。
Multifactor Explorationタブ
複数の実験因子を同時にPCA可視化に取り込むことができる。

PCA2GOタブ
PCA2GO タブでは、各主成分の負荷量の上下に表示される遺伝子のうち、生物学的な共通テーマが何かを理解することができる(pcaExplorerを追加パラメータなしで起動した時は利用不可)。
実行するには、アップロードした生物名と遺伝子IDの種類を指定する。間違っているとエラーになる。

出力例
Gene OntologyのBiological Processが使われる。topGOパッケージが使用されている。

他にもレポート生成機能などがあります。
引用
pcaExplorer: an R/Bioconductor package for interacting with RNA-seq principal components
Federico Marini, Harald Binder
BMC Bioinformatics. 2019; 20: 331
*1
大腸菌のシングルコロニー内でも、時間が経過すると発現プロファイルにバリエーションが出ることが報告されている(eLife)。バルクのタイムコースRNAseqと空間トランスクリプトーム解析でこれを明らかにしている。