研究者は、シングルおよびメタゲノムアセンブリの精度を客観的に評価し、それらに含まれる可能性のあるエラーを自動的に検出するための汎用的な手法を必要としている。現在の手法は、リファレンスを必要としたり、アセンブリ品質の多くの側面のうちの1つしか考慮していなかったり、統計的な正当性を欠いていたり、メタゲノムアセンブリを評価するように設計されていなかったりと、このニーズを十分に満たしていない。
この論文では、これらの限界を克服し、厳密な統計的手法を用いてリファレンスに依存しない方法でアセンブリの精度を体系的に評価するアセンブリ尤度評価(ALE)フレームワークを紹介している。このフレームワークは包括的なもので、リードの品質、メイトペアの向きとインサートの長さ(ペアエンドリードの場合)、シーケンスカバレッジ、リードアラインメント、k-mer頻度を統合している。ALEは、単一の塩基エラー、挿入/欠失、ゲノムリアレンジメント、メタゲノムで提示されるキメラアセンブリなど、単一および複数のメタゲノムアセンブリのsyntheticエラーをピンポイントで検出する。リアルデータのゲノムでは、ALEはSpirochaeta smaragdinaeの完成したゲノムから3つの大きなミスアセンブリを同定し、それらはすべてPacific Biosciencesのシーケンシングによって独立して検証された。イルミナデータを用いた一塩基レベルでは、GCリッチなRhodobacter sphaeroidesゲノムのトレーニングセットにおいて、222個の一塩基バリアントのうち215個(97%)を回収した。Pacific Biosciencesのリアルデータを使用した場合、ALEはLambda Phageゲノムの12個のsyntheticエラーのうち12個を特定し、Pacific Biosciences独自のバリアントコーラーEviConsをも上回った。要約すると、ALEフレームワークは、単一ゲノムおよびメタゲノムのアセンブリ精度を、基準に依存せず、統計的に厳密に測定する包括的な方法を提供する。ALEは、UoI/NCSAライセンスの下、オープンソースソフトウェアとして http://www.alescore.org で公開されている。CとPythonで実装されている。
Documentation
https://portal.nersc.gov/dna/RD/Adv-Seq/ALE-doc/
Assembly Likelihood Evaluationフレームワーク(ALE)は、リファレンスフリーのアプローチでゲノムおよびメタゲノムアセンブリを評価する。リードの品質、ペアエンドリードの向きとインサートサイズ、カバレッジ、リードアラインメント、k-mer頻度を用いる。ALEはベイズ統計を用いて2つの確率を定義する。(i)リード情報のないアセンブリの可能性を記述する確率分布、および(ii)アセンブリから生成されるリードの所定のセットの確率。これらの確率を組み合わせたものがALEスコア(アセンブリが正しい確率)であり、同じゲノムのアセンブリを比較する際に使用することができる。
(結果として得られるALEスコアは、同じゲノムの異なるアセンブリを比較するために使用することができるが、論文中で著者はALEを生物間のアセンブリの比較に使用すべきではないと述べている)
インストール
依存
git clone git@github.com:sc932/ALE.git
cd ALE/src/
make -j
#conda (link)
mamba install -c bioconda ale -y
> ./ALE
Welcome to the Assembly Likelihood Estimator!
(C) 2010 Scott Clark
Usage: ALE [-options] alignments.[s|b]am assembly.fasta[.gz] ALEoutput.txt
Options: <i>nt <f>loat <s>tring [default]
-h or --help : print out this help
--kmer <f> : Kmer depth for kmer stats [4]
--qOff <i> : Quality ascii offset (illumina) [33] or 64 (or 0)
--pl <s> : placementOutputBAM
--pm <s> : library parameter file (auto outputs .param)
--nout : only output meta information (no per base) [off]
--minLL : the minimum log Likelihood (-120)
--metagenome : Evaluate each contig independently for depth & kmer metrics
--realign[=matchScore,misMatchPenalty,gapOpenPenalty,gapExtPenalty,minimumSoftClip (default: 1,3,11,4,8) ]
Realign reads with Striped-Smith-Waterman honoring ambiguous reference bases
and stacking homo-polymer indels
for PacBio, try --realign=1,5,2,1,20 (similar to BWA-SW recommendations)
--SNPreport <s> : Creates a new text file reporting all SNP phasing
observed by a read against ambiguous bases in the reference
--minQual <i> : Minimum quality score to use in Z-normalization (default 3).
Illumina quality scores can be unreliable below this threshold
実行方法
1、評価したいリファレンス配列と、そのリファレンス配列にシークエンシングリードをマッピングして得たbam/samファイルを指定する。
ALE input.bam assembly.fasta.gz output.ale > log
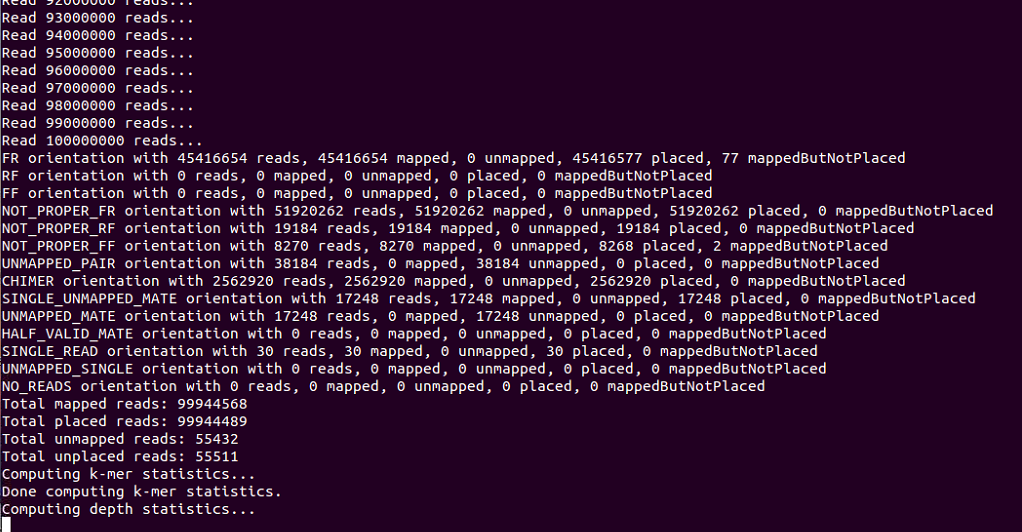
2、結果をwigファイルに変換する。
#python2コード
ale2wiggle.py ALEoutput.ale
#IGVを使わないならplotter3.pyを使用(環境が作れずに失敗した)
plotter3.py input.bam,ALEoutput.ale
IGVなどで視覚化できる。
引用
ALE: a generic assembly likelihood evaluation framework for assessing the accuracy of genome and metagenome assemblies
Scott C Clark, Rob Egan, Peter I Frazier, Zhong Wang
Bioinformatics. 2013 Feb 15;29(4):435-43
関連