2018 10/6 タイトル修正
2019 説明修正、dockerリンク追加、インストール方法追加、dockerリンク修正 、コマンド修正、help追加、タイトル修正、インストールの説明の誤り修正、バージョンアップ追記、crisper、IS、AMR tag追加、誤字修正、dockerを使う例を追記
2020 4/19 multiqcとの連携例、4/28 コマンド例追記、10/ 5 help更新、10/20 追記、12/28 コマンド例追加
2021 5/9 condaのインストール手順アップデート、 9/25 インストール手順修正
2023/01/14 mt追記、07/04 virus追記, 10/19 追記
2024/06/02 --cpus 16追加
Prokkaは、バクテリア、アーキア、ウィルスのアノテーションツール。はじめにblast+でcore geneを特定し、それからHMMER3を使ってより高感度かつ精度の高いコード領域の特定が行われる。
Prokka passed 10,000 citations on Google Scholar this week. It's mind-blowing to think it has annotated millions of bacterial genomes over the last 8 years. Thanks to everyone for your support and feedback. 💚🦠🧬💻 pic.twitter.com/kKfKs1vjAe
— Torsten Seemann (@torstenseemann) December 26, 2022
バージョン
Releases · tseemann/prokka · GitHub
インストール
ubuntu18.0.4のpython3.9環境(mambaforge3.9)にてテストした。
本体 Github
conda、またはbrewで導入する。
#bioconda (link) (python3.8でもインストール可能) ここでは高速なmambaを使用
mamba install -c conda-forge -c bioconda -c defaults prokka -y
#python3のコードだが、condaで入れる際に依存するperlライブラリがなぜか導入されず。prokkaを動かせない場合がある。試した際は3.8で失敗し、3.6では全てのライブラリが正常に導入された。
mamba create -n prokka
conda activate prokka
mamba install -c conda-forge -c bioconda -c defaults prokka -y
#エラーが起こったので導入方法を変更(ubuntu18のdockerイメージでは正しく導入された)
mamba create -n prokka_env python=3.7 -y
conda activate prokka_env
mamba install -c bioconda -y perl-bioperl==1.7.2
mamba install -c bioconda prokka -y
#homebrew
brew install prokka
#perlライブラリが入らなければcpanmで導入 -Lでパス指定
sudo cpan Time::Piece XML::Simple Digest::MD5 Bio::Perl
#インストールができない場合、gitで最新版をダウンロードする方法が推奨されている
git clone https://github.com/tseemann/prokka.git
cd prokka/ #ダウンロードしたprokka/に移動
bin/prokka --setupdb #Index the sequence databasesをする
dockerイメージ
https://hub.docker.com/r/staphb/prokka/
docker pull staphb/prokka:latest
> prokka -h
# prokka
Name:
Prokka 1.14.5 by Torsten Seemann <torsten.seemann@gmail.com>
Synopsis:
rapid bacterial genome annotation
Usage:
prokka [options] <contigs.fasta>
General:
--help This help
--version Print version and exit
--citation Print citation for referencing Prokka
--quiet No screen output (default OFF)
--debug Debug mode: keep all temporary files (default OFF)
Setup:
--dbdir [X] Prokka database root folders (default '/prokka-1.14.5/db')
--listdb List all configured databases
--setupdb Index all installed databases
--cleandb Remove all database indices
--depends List all software dependencies
Outputs:
--outdir [X] Output folder [auto] (default '')
--force Force overwriting existing output folder (default OFF)
--prefix [X] Filename output prefix [auto] (default '')
--addgenes Add 'gene' features for each 'CDS' feature (default OFF)
--addmrna Add 'mRNA' features for each 'CDS' feature (default OFF)
--locustag [X] Locus tag prefix [auto] (default '')
--increment [N] Locus tag counter increment (default '1')
--gffver [N] GFF version (default '3')
--compliant Force Genbank/ENA/DDJB compliance: --addgenes --mincontiglen 200 --centre XXX (default OFF)
--centre [X] Sequencing centre ID. (default '')
--accver [N] Version to put in Genbank file (default '1')
Organism details:
--genus [X] Genus name (default 'Genus')
--species [X] Species name (default 'species')
--strain [X] Strain name (default 'strain')
--plasmid [X] Plasmid name or identifier (default '')
Annotations:
--kingdom [X] Annotation mode: Archaea|Bacteria|Mitochondria|Viruses (default 'Bacteria')
--gcode [N] Genetic code / Translation table (set if --kingdom is set) (default '0')
--prodigaltf [X] Prodigal training file (default '')
--gram [X] Gram: -/neg +/pos (default '')
--usegenus Use genus-specific BLAST databases (needs --genus) (default OFF)
--proteins [X] FASTA or GBK file to use as 1st priority (default '')
--hmms [X] Trusted HMM to first annotate from (default '')
--metagenome Improve gene predictions for highly fragmented genomes (default OFF)
--rawproduct Do not clean up /product annotation (default OFF)
--cdsrnaolap Allow [tr]RNA to overlap CDS (default OFF)
Matching:
--evalue [n.n] Similarity e-value cut-off (default '1e-09')
--coverage [n.n] Minimum coverage on query protein (default '80')
Computation:
--cpus [N] Number of CPUs to use [0=all] (default '8')
--fast Fast mode - only use basic BLASTP databases (default OFF)
--noanno For CDS just set /product="unannotated protein" (default OFF)
--mincontiglen [N] Minimum contig size [NCBI needs 200] (default '1')
--rfam Enable searching for ncRNAs with Infernal+Rfam (SLOW!) (default '0')
--norrna Don't run rRNA search (default OFF)
--notrna Don't run tRNA search (default OFF)
--rnammer Prefer RNAmmer over Barrnap for rRNA prediction (default OFF)
実行方法
アセブルして作ったscaffodls.fastaやFinished genomeをアノテーションする(*1)。16スレッド指定。
prokka genome.fa -o prokka_output --cpus 16
ランが終わるとoutputディレクトリの中にgtf、gff、faa、fnn、gbfなどができる。gbfがgenbank形式のファイルになる。

データベースに登録するためのオプションや、Genus特異的に調べるオプションもある。またローカルにすでにたくさんのgenbankファイルを持っている人なら、それをデータベースにして、精度の高いアノテーション解析を行うことができる。詳細は公式マニュアルを確認してください。
出力ファイル名のprefixを指定、デフォルトはprokka+日付だがEcoliに変更。
prokka genome.fa -o prokka_output --prefix Ecoli --cpus 16
locus tag名をJ001に変更。
prokka genome.fa -o prokka_output --prefix Ecoli --locustag J001 --cpus 16
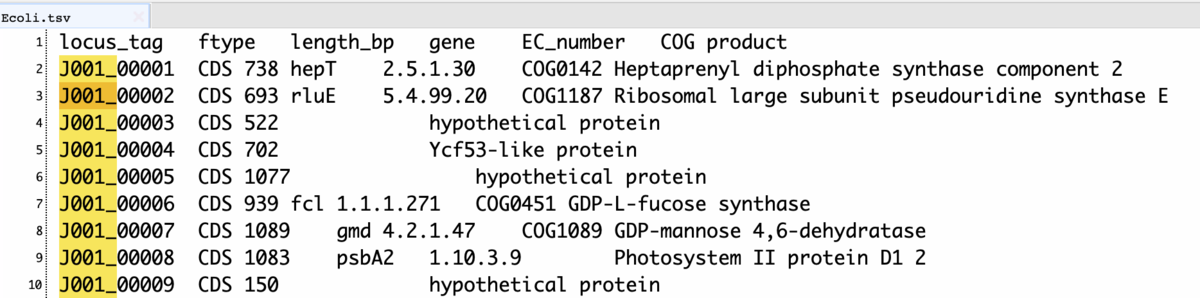
あとでアノテーションを追加することを想定して、番号を1ずつ増加から5きざみに変更。
prokka genome.fa -o prokka_output --prefix Ecoli --locustag J001 --increment 5 --cpus 16
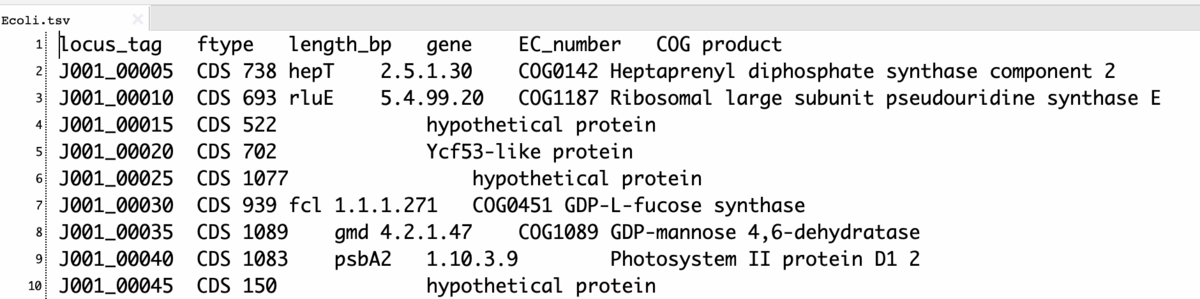
上記に加え、GFFのバージョンは3、アノテーションバージョンは1、センター名はcenter_of_genomicsを追加。また、デフォルトのCDSに加えてgene(--addgenes)と mRNA(--addmrna)をアノテーションフィーチャーとして追加。
prokka genome.fa -o prokka_output --prefix Ecoli --locustag J001 --increment 5 --gffver 3 --centre center_of_genomics --addgenes --addmrna --cpus 16

metagenome(binningしていない複数ゲノムが混ざった配列、binning後の品質の高いMAGなら--metagenomeは付けない)
prokka assembly.fasta -o prokka_output --metagenome --cpus 16
- --metagenome Improve gene predictions for highly fragmented genomes (default OFF)
ミトコンドリアゲノム。codon tableを変更するか(参考)、kingdomオプションをつける。あとでアノテーションを追加することを想定して、番号を1ずつ増加から5きざみに変更。
prokka genome.fa -o prokka_output --prefix mitogenome --locustag mt --increment 5 --kingdom Mitochondria
コメント;種によってはイントロンが存在するミトコンドリア遺伝子もあり、そのような遺伝子ではexon-intron境界がずれていたり、一部のexonしか予測できなかったりする。rRNAにも遺伝子とは別のタイプのintronが存在することがあり、rRNAも注意が必要(rRNAなら近縁種とのblastn検索に基づく手動アノテーションがより正確)。あくまでprokkaはイントロンがない原核生物向けの遺伝子予測ツールであり、遺伝子予測のhintにしかならないことに注意したい。
prokkaの代わりに真核生物向けのab initio遺伝子予測手法が有効ではないかと考えるかもしれない。しかし、ab initio予測ツールの訓練にはintronを持つある程度たくさんの遺伝子が必要であり、サイズが小さいmtで十分な訓練ができるかどうかは疑わしい。また、自動アノテーションを行うwebサーバーもいくつか公開されているが、モデル生物から遠い場合、精度が低い可能性がある。これらを考慮すると、mtゲノムの決まった種から進化的に遠いmitogenomeのアノテーションでは、保存されたタンパク質のマッピングなどに基づくマニュアルのアノテーションが早いかもしれない。手動で進めるならこちらが役立つ。RNA-seqのイントロンを考慮したマッピング結果もヒントの1つに使える。ESTやTrinityのassembly(TransDecorderで選抜した配列)はgmap(紹介)などでアラインできる。保存されたタンパク質のマッピングにはexonerateなどが使える。prokkaの遺伝子モデルも参考になる。既存の分類群から遠いウィルスも同様と思われる。
ウィルスゲノム。
prokka genome.fa -o prokka_output --prefix prefix --locustag LOCUS --increment 5 --kingdom Viruses
1、ゲノムサイズが小さいと訓練が不十分になり、精度が上がらない可能性がある。アノテーション後、ORFが予測されていない領域をORF finderでチェックする。予測されたタンパク質産物はBLASTpサーチしてウィルスで予測されるタンパク質が出てくるのかなど確認する。後で十分な品質チェックを行うなら利用可能と考えられる。
2、環状アセンブリでは、アノテーション時に見かけの開始点にORFがないか注意する。oriがわかっているならその上流を+1にする。分からないなら、一度アノテーションを行なってORFが見つからない領域を探す。そのような領域を見かけ上の+1にするのが無難と思われる。
dockerを使う。
docker run --rm -itv $PWD:/data/ -w /data staphb/prokka:latest \
prokka input_genome.fasta -o prokka_output
2020 4/19 追記、2020 12/28オプション追加
multiqcと連携する。
以下のスクリプトでディレクトリ中の全fastaのアノテーションを行う。gene IDは入力fasta名から取る(--locustag $folder)。出力ディレクトリ(--prefix $folder)と株名も同様(--strain $folder)。
#!/bin/sh
#prooka annotation
for file in `\find *fasta -maxdepth 1 -type f`; do
genome=${file}
folder=${file%.fasta}
prokka $genome --outdir $folder --addgenes --prefix $folder --strain $folder --cpus 12 --quiet --locustag $folder --prefix $folder
done
prokka.shとして保存。--strain xxx がmultiqcで出力される名前になる。
実行。
bash prokka.sh
multiqc .
2020 4/20 追記
PROKKAで出力されるgffからbedを出力。
#BEDOPSを入れていないなら導入
apt insall BEDOPS
convert2bed --input=gff < PROKKA.gff > PROKKA.bed
2021 11/3
genetic codeはデフォルトでは細菌、アーキアなどの11になっています(--kingdom Bacteriaがデフォルトで、これにより--gcodeのデフォルトである0から11に変更されている)。
参考ページ
引用
Prokka: rapid prokaryotic genome annotation
Seemann T
Bioinformatics. 2014 Jul 15;30(14):2068-9
2023/03/29
prokkaはコンティグ末端のORF予測で、不完全なものを除外する可能性があります。 そのため、コンティグの連続性が低い場合、コンティグ末端のORF予測を排除することで、そのような遺伝子がコア遺伝子探索でMissingになる可能性があります。自分の場合はprokkaのベースとなるprodigalに変えることで予測できるようになりました。再現性がない現象かもしれませんが注意してください。
(ただしprodigalの出すGFF3は微妙にフォーマットがおかしく、そのままパンゲノム解析などに使用するとエラーになる。prokkaの出す標準化されたGFF3は貴重)
*1
tbl2asnでエラーになったら、tbl2asnだけ導入し直してください。NCBIの定めた使用期限が設定されているようです。
Could not run command: tbl2asn · Issue #139 · tseemann/prokka · GitHub
関連
2019/08
バージョンアップされてますね。
Everyone stop everything and update your Prokka version! Looks like v1.14.0 was just released 4 days ago! New and improved - Now with AMR and IS databases! Do I sound like an infomercial salesman?!https://t.co/hWgI31yLkN
— Taj Azarian (@EpiDemos82) August 12, 2019