最新のショートリードシーケンサーからの出力が増加したことにより、ローカバレッジホールゲノムシーケンス(LC-WGS)は大規模な系統学へのアプローチとしてますます手頃な価格になっている。しかし、従来のシーケンシング手法と比較していくつかの利点があるにもかかわらず、このタイプのデータを系統学的に処理するためのツールはほとんど存在しない。LC-WGSゲノムは断片化されているため、オルガネラゲノムやリボソーム遺伝子のような組み立てが容易でコピー数が多い領域に限定して使用されてきた。本発表では、アセンブルされたゲノムから系統樹マーカーを直接マイニングする新しい方法を紹介する。まず、アラインメント検索により相同領域を求め、次に「ヒットスティッチング」によって隣接あるいは重複する領域を連結していく。最後に、新しいスライディングウィンドウ技術により、アラインメントから非コード領域を切り出す。環形動物Dimorphilus gyrociliatusにおけるnear-universal single-copy orthologs (USCOs)をリカバリーすることにより、Patchworkの有用性を実証する。
マニュアルより
全ゲノム配列決定(WGS)はますます安価になってきていますが、このようなデータを扱うための適切なソフトウェアツールがないため、このアプローチは通常、系統研究にあまり利用されてきませんでした。WGSをゲノムスケールの系統解析に利用することで、分類群サンプリングやデータの再利用性が劇的に向上する可能性があり、生成されたデータは多くの異なるタイプの解析に利用できる可能性があります。このような背景から、私たち(著者ら)はWGSデータから直接系統樹マーカーを検索する新しいプログラム、Patchworkを設計することにしました。
Patchworkは、全ゲノムシーケンス(WGS)データから系統樹マーカーを検索し、連結するためのアライメントベースのプログラムです。このプログラムは、提供されたDNAのコンティグ(塩基配列)を1つまたは複数のアミノ酸参照配列と照合します。複数のアミノ酸配列がヒットした場合、それぞれのアミノ酸配列に対して1つの連続した配列が作成されます。
- 塩基配列とタンパク質配列のアラインメントを行う。
- 重なり合った配列やギャップある配列を参照配列に基づいてつなぎ合わせる。
- 遠縁の分類群であってもホモログを見つけることが可能。
- Juliaで記述され、DIAMONDを利用して最高速度を実現。
- グラフィカルオーバービュー。
We think there is unrealised potential of genome skimming approaches for phylogenomics and blame it on a lack of appropriate tools. We came up with Patchwork (https://t.co/Spx06YVHDv) for mining markers in a fast and user-friendly way. Very excited to tell you more soon! pic.twitter.com/HGFPygD2IN
— Felix Thalén (@fethalen) April 7, 2022
https://github.com/fethalen/Patchwork/wiki
インストール
docker imageを使ってテストした。
#依存
mamba create -n patchwork -y
conda activate patchwork
mamba install -c conda-forge julia -y
mamba install -c bioconda diamond -y
#本体のビルド
git clone https://github.com/fethalen/patchwork
cd patchwork/
julia Patchwork.jl --help
#conda #作業進行中。現在はまだ対応していないので注意。
mamba install -c bioconda patchwork -y
#dockerhub
docker pull biocontainers/patchwork:v0.1.2_cv1
> patchwork -h
usage: <PROGRAM> --contigs PATH [PATH...] --reference PATH [PATH...]
[--output-dir PATH] [--diamond-flags LIST]
[--makedb-flags LIST] [--matrix NAME]
[--custom-matrix PATH] [--gapopen NUMBER]
[--gapextend NUMBER] [--threads NUMBER]
[--species-delimiter CHARACTER]
[--wrap-column NUMBER] [--overwrite] [--version] [-h]
Alignment-based retrieval and concatenation of phylogenetic markers
format whole-genome sequencing (WGS) data
optional arguments:
--contigs PATH [PATH...]
Path to one or more sequences in FASTA format
--reference PATH [PATH...]
Either (1) a path to one or more sequences in
FASTA format, (2) a subject
database (DIAMOND or BLAST database), or (3) a
DIAMOND output file in
tabular format.
--output-dir PATH Write output files to this directory (default:
"patchwork_output")
--diamond-flags LIST Flags sent to DIAMOND (type: Vector{String},
default: ["--ultra-sensitive"])
--makedb-flags LIST Flags sent to DIAMOND makedb (type:
Vector{String})
--matrix NAME Set scoring matrix
--custom-matrix PATH Use a custom scoring matrix
--gapopen NUMBER Set gap open penalty (positive integer) (type:
Int64)
--gapextend NUMBER Set gap extension penalty (positive integer)
(type: Int64)
--threads NUMBER Number of threads to utilize (default: all
available) (type: Int64, default: 128)
--species-delimiter CHARACTER
Used to distinguish the OTU from the rest in
sequence IDs (type: Char, default: '@')
--wrap-column NUMBER Wrap output sequences at this column number
(default: no wrap) (type: Int64, default: 0)
--overwrite Overwrite output from previous runs without
warning
--version show version information and exit
-h, --help show this help message and exit
テストラン
patchworkをランするには、--contigsでDNA配列を含むFASTAファイルのパスを、--referenceでアミノ酸配列を含むFASTAファイルのパスを指定する。
git clone https://github.com/fethalen/Patchwork.git
cd Patchwork/test/
sudo docker run --rm -itv $PWD:/data -w /data --user $(id -u) biocontainers/patchwork:v0.1.2_cv1
> patchwork --contigs 07673_dna.fa --reference 07673_Alitta_succinea.fa --output-dir patchwork_output
- --contigs Path to one or more sequences in FASTA format
- --reference Either (1) a path to one or more sequences in FASTA format, (2) a subject database (DIAMOND or BLAST database), or (3) a DIAMOND output file in tabular format.
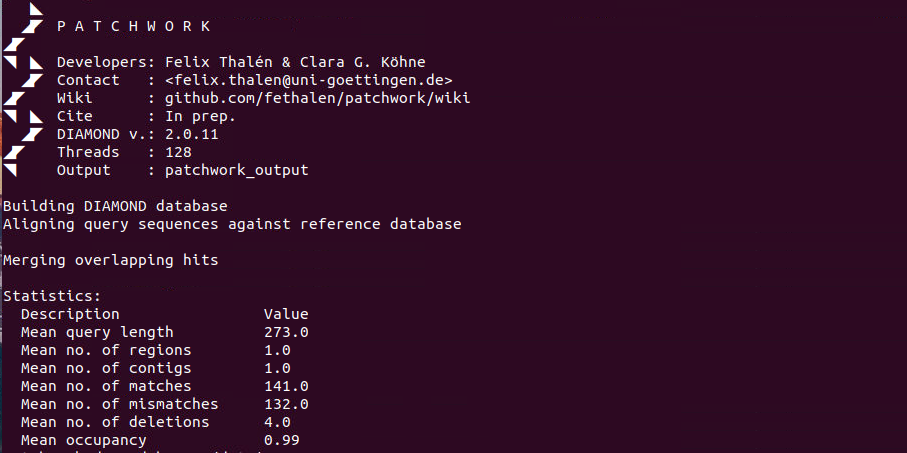
出力
patchwork_output/

- alignments.txt には、各アラインメントの視覚的な表現が含まれている。
- database.dmndは、DIAMONDで作成されたデータベースファイル。
- diamond_blastx.logには、DIAMONDの出力がプレーンテキストで格納されている。
- diamond_makedb.logには、データベース構築のためのDIAMONDの出力がプレーンテキストで格納されている。
- diamond_resultsには、各参照配列の結果が格納されている。
- queries_outには、マージされ、翻訳されたFASTA形式のクエリー配列が格納されている。
- statistics には、個々の検索結果(statistics/statistics.csv)およびすべての検索結果(statistics/average.csv)に対する基本統計値が含まれる。
引用
Patchwork: alignment-based retrieval and concatenation of phylogenetic markers from genomic data
Felix Thalen, Clara Gisela Koehne, Christoph Bleidorn
bioRxiv, Posted July 03, 2022
関連
参考
”ゲノムスキミングとは、ゲノムをローパスで浅く(最大5%)シーケンスし、ゲノムスキムと呼ばれるDNA断片を生成するシーケンス手法である。 ゲノムの高コピー部分とは、リボソームDNA、プラスミドゲノム(プラストム)、ミトコンドリアゲノム(ミトゲノム)、マイクロサテライトやトランスポゾームなどの核内リピートから構成されている。 これらのゲノムスキミングは「ゲノム氷山の一角」に過ぎないが、系統樹解析により、従来の手法よりも低コストかつ大規模に進化史や生物多様性に関する知見を得ることができる。 ゲノムスキミングに必要なDNA量が少ないため、その手法はゲノム以外の分野にも適用可能である。”
Genome skimming
https://en.wikipedia.org/wiki/Genome_skimming