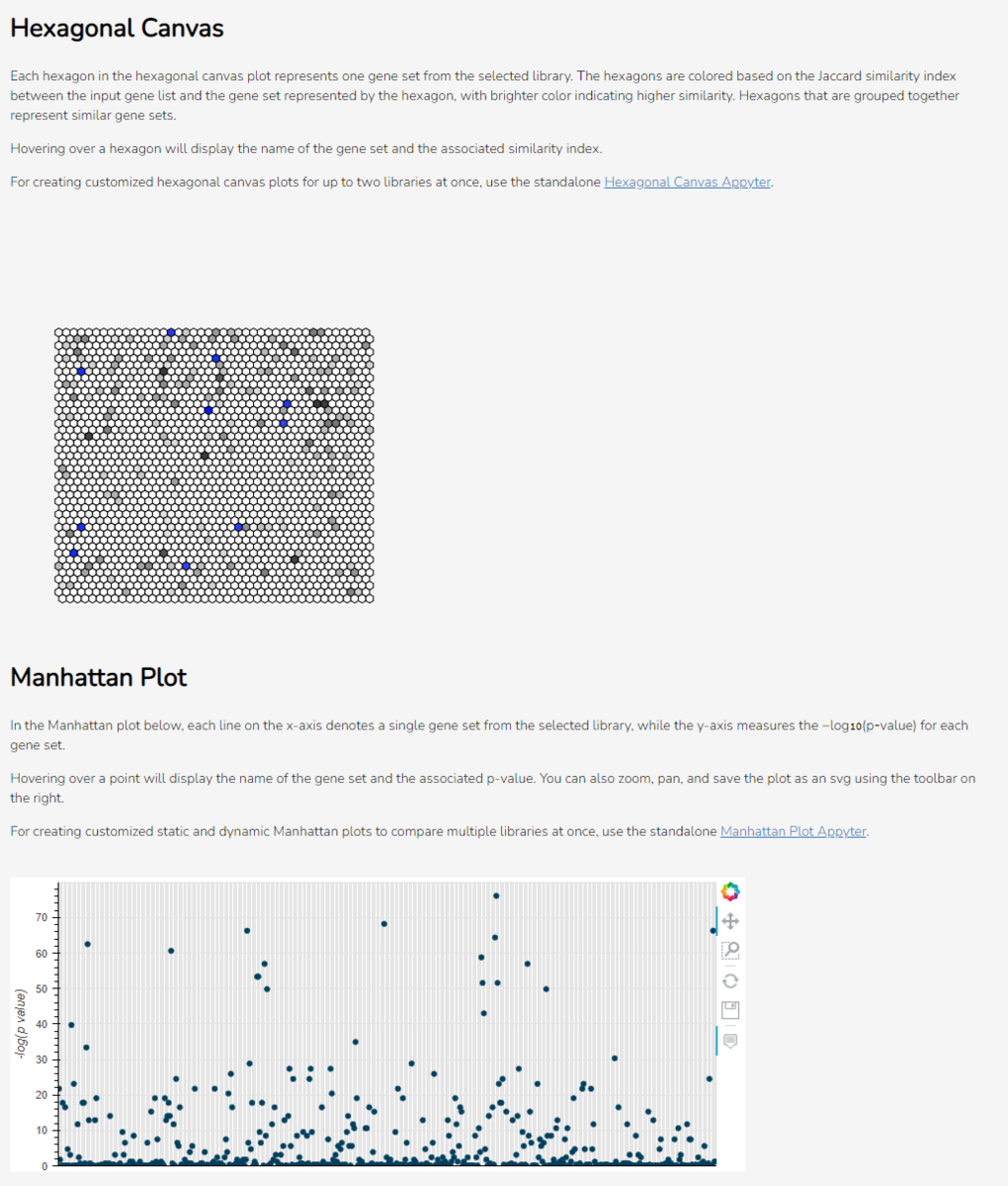
Minhashは、2つの集合の類似性をJaccard指数(集合の和に対する交点の大きさの比として定義される)の観点から推定する確率的な手法である。この手法は、対象となる集合の大きさが似ている場合に最も優れた性能を発揮し、集合の大きさが大きく異なる場合には性能が大幅に低下することを実証した。本論文では、大きさの異なる集合のJaccard indexを推定するのに適した、「containment minhash approach」と呼ばれる新しい効率的な手法を紹介する。これは、高速なメンバーシップクエリのための別の確率的手法(特にBloomフィルタ)を利用することで実現している。containment Minhash法の推定誤差の確率を導出し、古典的なMinhash法を大幅に改善することを示す。また、時間的・空間的な複雑さの点でも大きな改善が見られる。応用例として、この手法を用いてメタゲノムデータセット中の生物の有無を検出し、非常に小さく、存在量の少ない微生物の存在を検出できることを示す。
インストール
mamba install -c bioconda cmash -y
> MakeDNADatabase.py -h
usage: MakeDNADatabase.py [-h] [-p PRIME] [-t THREADS] [-n NUM_HASHES]
[-k K_SIZE] [-i]
in_file out_file
This script creates training/reference sketches for each FASTA/Q file listed
in the input file.
positional arguments:
in_file Input file: file containing (absolute) file names of
training genomes.
out_file Output training database/reference file (in HDF5
format)
optional arguments:
-h, --help show this help message and exit
-p PRIME, --prime PRIME
Prime (for modding hashes) (default: 9999999999971)
-t THREADS, --threads THREADS
Number of threads to use (default: 128)
-n NUM_HASHES, --num_hashes NUM_HASHES
Number of hashes to use. (default: 500)
-k K_SIZE, --k_size K_SIZE
K-mer size (default: 21)
-i, --intersect_nodegraph
Optional flag to export Nodegraph file (bloom filter)
containing all k-mers in the training database. Saved
in same location as out_file. This is to be used with
QueryDNADatabase.py (default: False)
~
~
~
> MakeNodeGraph.py -h
usage: MakeNodeGraph.py [-h] [-fp FP_RATE] [-i INTERSECT_NODEGRAPH]
[-k K_SIZE] [-t THREADS]
in_file out_dir
This script will create node graph for a given k-mer size and query file (can
be used as input to QueryDNADatabase.py)
positional arguments:
in_file Input file: FASTQ/A file (can be gzipped).
out_dir Output directory
optional arguments:
-h, --help show this help message and exit
-fp FP_RATE, --fp_rate FP_RATE
False positive rate. (default: 0.0001)
-i INTERSECT_NODEGRAPH, --intersect_nodegraph INTERSECT_NODEGRAPH
Location of Node Graph. Will only insert query k-mers
in bloom filter if they appear anywhere in the
training database. Note that the Jaccard estimates
will now be J(query intersect union_i training_i,
training_i) instead of J(query, training_i), but will
use significantly less space (unfortunately will also
disable threading). (default: None)
-k K_SIZE, --k_size K_SIZE
K-mer size (default: 21)
-t THREADS, --threads THREADS
Number of threads to use (default: 128)
> QueryDNADatabase.py -h
usage: QueryDNADatabase.py [-h] [-t THREADS] [-f] [-fp FP_RATE]
[-ct CONTAINMENT_THRESHOLD] [-c CONFIDENCE]
[-ng NODE_GRAPH] [-b] [-i]
in_file training_data out_csv
This script creates a CSV file of similarity indicies between the input file
and each of the sketches in the training/reference file.
positional arguments:
in_file Input file: FASTQ/A file (can be gzipped).
training_data Training/reference data (HDF5 file created by
MakeTrainingDatabase.py)
optional arguments:
-h, --help show this help message and exit
-t THREADS, --threads THREADS
Number of threads to use (default: 128)
-f, --force Force creation of new NodeGraph. (default: False)
-fp FP_RATE, --fp_rate FP_RATE
False positive rate. (default: 0.0001)
-ct CONTAINMENT_THRESHOLD, --containment_threshold CONTAINMENT_THRESHOLD
Only return results with containment index above this
value (default: 0.02)
-c CONFIDENCE, --confidence CONFIDENCE
Desired probability that all results were returned
with containment index above threshold [-ct] (default:
0.95)
-ng NODE_GRAPH, --node_graph NODE_GRAPH
NodeGraph/bloom filter location. Used if it exists; if
not, one will be created and put in the same directory
as the specified output CSV file. (default: None)
-b, --base_name Flag to indicate that only the base names (not the
full path) should be saved in the output CSV file
(default: False)
-i, --intersect_nodegraph
Option to only insert query k-mers in bloom filter if
they appear anywhere in the training database. Note
that the Jaccard estimates will now be J(query
intersect union_i training_i, training_i) instead of
J(query, training_i), but will use significantly less
space. (default: False)
実行方法
リファレンス/トレーニングデータベースを作成し、ブルームフィルターを形成し、データベースに問い合わせるという流れで使う。
1、fasta/fastqのフルパスを指定した1行ずつ指定したファイルを指定してデータベースを作成。
MakeDNADatabase.py list.txt TrainingDatabase.h5
2、(任意)ブルームフィルタを作成
MakeNodeGraph.py TrainingDatabase.h5 outdir
3、クエリファイルMetagenome.faとデータベースとのJaccard indexの推定値を得る
QueryDNADatabase.py Metagenome.fa TrainingDatabase.h5 output.csv
引用
Improving MinHash via the containment index with applications to metagenomic analysis
David Koslickia, Hooman Zabetib
https://doi.org/10.1016/j.amc.2019.02.018
関連