2023/08/11 説明を修正
2024/08/20 追記, kraken1 からkraken2に変更
GTDBでもサードパーティとして紹介されているが、レポジトリGTDB_KrakenでGTDBのリリースR86のkrakenデータベースが公開されている(属レベルでアサインされていない分類 (g__) は排除されている)。ビルド済みなので、ダウンロードしてkraken1とkraken2で直ぐに使用できる。
https://drive.google.com/file/d/18E0W_ezNLAhxxZjjelQYwoLA_0oBm4Lo/view?usp=sharing
解凍して出来たディレクトリを指定してkrakenをランする。
kraken2 --db kraken2-full-database --threads 20 --paired --report kraken2_report.txt --use-names --gzip-compressed --report-zero-counts --classified-out seqs#.fq sample_R1.fq.gz sample_R2.fq.gz > output.txt
このGTDB_Krakenレポジトリでは、ソースからkraken D.Bをビルドするスクリプトも公開されている。これは、Entrez Directを使ってGTDB R86に対応するゲノム配列とtaxonomy情報をNCBIからダウンロードし、kraken-buildコマンドを使ってkraken D.Bをビルドするスクリプトとなる。このスクリプトの使い方を確認しておきます。
インストール
ソースからkraken GTDB R86 DBをビルドするには、レポジトリをcloneし、配置されているリストファイル”gtdb.2018-12-10.tsv”を指定してperlスクリプト:gtdbToTaxonomy.plをランする。
依存
- Edirect - https://www.ncbi.nlm.nih.gov/books/NBK179288/
- Perl
- BioPerl - https://bioperl.org/INSTALL.html
- Kraken or Kraken2
git clone https://github.com/hcdenbakker/GTDB_Kraken.git
cd GTDB_Kraken/
テストラン
まずテストランを行う。依存関係は先に導入しておく。kraken2ではなくkraken1が必要。
cd GTDB_Kraken/tests/
chmod +x 01_Salmonella.sh
bash 01_Salmonella.sh
この小さなテストランでは、GTDB R86の"g__Salmonella"に限定してゲノム配列をダウンロードし、kraken DBをビルドする。
gtdb.2018-12-10.tsvは、R86のゲノムのアクセッションIDとGTDBの分類が1行ずつ書かれたされたタブ区切りのリスト。”RS_GCF_001300075.1”のように、GTDB系統樹のtipラベルは"RS_"または "GB_"で始まり、その後にNCBIのGenBankかRefSeqアセンブリのアクセッションIDが続く。タブで区切られ、2列目にはGTDBのfull taxonomic lineageが書かれている。

1,01_Salmonella.shスクリプトは、このリストからgrepで g__Salmonellaを含む行だけを抽出する。
grep g__Salmonella ../data/gtdb.2018-12-10.tsv > Salmonella_list
2,抽出したリストを指定してperl スクリプトgtdbToTaxonomy.plを実行している。
perl ../scripts/gtdbToTaxonomy.pl --infile Salmonella_list
#リストのfastaはダウンロードされてgtdb/に保存される。もしローカルのfastaファイルを使用するなら--sequence-dirオプションを立ててローカルのgenome dirを指定する。当然listのファイル名と一致していないといけない。
perl ../scripts/gtdbToTaxonomy.pl --infile Salmonella_list --sequence-dir genome_dir/
このスクリプトにより、2つのディレクトリができ、さらにgtdbディレクトリが出来て、その中にfastaファイルがダウンロードされる。--sequence-dirを付けた時は指定したローカルストレージのディレクトリからgtdb/にコピーされる。

1つ目のtaxnomy/にはnames.dmpとnodes.dmpが含まれる。

names.dmpはtaxIDとその分類階級および学名とのマッピング情報を提供する。NCBIで使用されているものと同じフォーマットでなければならない(参考)。\t|\tがセパレータ。
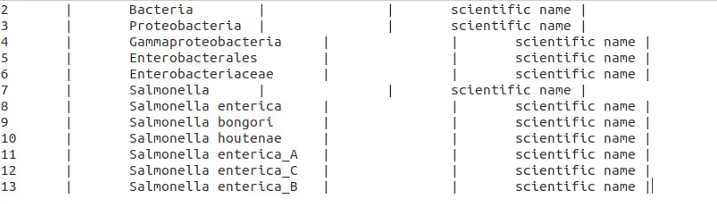
nodes.dmpは各taxonのID、親taxonのID、階層レベルなどが含まれている。やはりNCBI標準のnodes.dmpと同じでなければならない。\t|\tがセパレータ。

数値の意味などの詳細はこちら;https://ftp.ncbi.nlm.nih.gov/pub/taxonomy/taxdump_readme.txt
2つ目のlibrary/gtdb/にはダウンロードされたゲノムのfastaファイルが含まれる。

3、この2つのディレクトリからkraken DBが作成される。まずゲノムを追加し、それからビルドする。
for i in library/gtdb/*.fna; do
kraken2-build --add-to-library $i --db GTDB_Kraken
done
#2 GTDB_Kraken/にtaxonomy/を移動させる。
mv taxonomy GTDB_Kraken/
#3 build
kraken2-build --build --db GTDB_Kraken
下の画像がテストランの最終成果物。
GTDB_Kraken/

上のコマンドは通常のkraken DBを作成する時と同じ流れで進められている。
#通常のkraken DB作成フロー(全自動の”standard”もある)
#1 まずNCBI taxonomy database dumpをダウンロード
kraken2-build --download-taxonomy --db kraken2_DB
=> kraken2_DB/下にtaxonomyディレクトリができ、Taxonomy database dump(taxdmp)のセットがダウンロードされる。nodes.dmpやnames.dmpも含まれる
#2 続いてRefSeqリファレンスゲノムをダウンロード。いくつか選択できる(manual)。viralなら(600MBくらい)
kraken2-build --download-library viral --db kraken2_DB
#plantなら(140GBくらい)
kraken2-build --download-library plant --db kraken2_DB
=> kraken2_DB/下のlibrary/viral/にゲノムがダウンロードされる。複数も可能(*1画像)
#3 さらにゲノムを追加できる。認識できるfastaのヘッダにルールがあるので注意(manual)
kraken2-build --threads 4 --add-to-library $file --db kraken2_DB
#4 最後にbuild。k値の指定など感度と精度に影響するオプションや、Low-complexity配列マスクなどのオプションがある(デフォルトON)
kraken2-build --threads 20 --build --db kraken2_DB
=> kraken2_DB/下に複数のファイルができる。ビルド後はkraken2_DB/の配列などは削除してOK
Kraken DB作成手順を一言で述べると、taxdmpファイル群とゲノムをダウンロードし、ビルドするという流れ。
GTDB_Krakenの実行方法
流れはテストランと全く同じ。ゲノムが多いので、ダウンロードにはかなりの時間がかかる。
#1
cd GTDB_Kraken/
perl scripts/gtdbToTaxonomy.pl --infile data/gtdb.2018-12-10.tsv
#すでにローカルにgenome.fastaがあるなら --sequence-dirオプションでそのディレクトリを指定する。サブディレクトリ以下も検索するのでディレクトリ直下にfastaファイルがなくてもOK
perl ../scripts/gtdbToTaxonomy.pl --infile data/gtdb.2018-12-10.tsv --sequence-dir local_GTDB_db_dir/
#2
db=GTDB_Kraken
for i in library/gtdb/*.fna; do
kraken-build --add-to-library $i --db $db
done
mv -v taxonomy $db
#3 $db/以下にtaxonomyとlibraryディレクトリを配置して以下のコマンドでビルド(kraken1)
kraken-build --build --db $db --threads 20
時間はかかるが、リストは6192行しかないので1~2日待っていれば終わる(テスト時は1日半かかった)。リクエストが多すぎるというエラーが出るときはレポジトリ参照。
引用
GitHub - hcdenbakker/GTDB_Kraken: Kraken(2) database based on the GTDB project
参考
Phyloplus
https://phylo.jifsan.org/files/Supplementary_Information.pdf
*1画像

kraken2 help
#help
> kraken2-build
$ kraken2-build
Must select a task option.
Usage: kraken2-build [task option] [options]
Task options (exactly one must be selected):
--download-taxonomy Download NCBI taxonomic information
--download-library TYPE Download partial library
(TYPE = one of "archaea", "bacteria", "plasmid",
"viral", "human", "fungi", "plant", "protozoa",
"nr", "nt", "UniVec", "UniVec_Core")
--special TYPE Download and build a special database
(TYPE = one of "greengenes", "silva", "rdp")
--add-to-library FILE Add FILE to library
--build Create DB from library
(requires taxonomy d/l'ed and at least one file
in library)
--clean Remove unneeded files from a built database
--standard Download and build default database
--help Print this message
--version Print version information
Options:
--db NAME Kraken 2 DB name (mandatory except for
--help/--version)
--threads # Number of threads (def: 1)
--kmer-len NUM K-mer length in bp/aa (build task only;
def: 35 nt, 15 aa)
--minimizer-len NUM Minimizer length in bp/aa (build task only;
def: 31 nt, 12 aa)
--minimizer-spaces NUM Number of characters in minimizer that are
ignored in comparisons (build task only;
def: 7 nt, 0 aa)
--protein Build a protein database for translated search
--no-masking Used with --standard/--download-library/
--add-to-library to avoid masking low-complexity
sequences prior to building; masking requires
dustmasker or segmasker to be installed in PATH,
which some users might not have.
--max-db-size NUM Maximum number of bytes for Kraken 2 hash table;
if the estimator determines more would normally be
needed, the reference library will be downsampled
to fit. (Used with --build/--standard/--special)
--use-ftp Use FTP for downloading instead of RSYNC; used with
--download-library/--download-taxonomy/--standard.
--skip-maps Avoids downloading accession number to taxid maps,
used with --download-taxonomy.
--load-factor FRAC Proportion of the hash table to be populated
(build task only; def: 0.7, must be
between 0 and 1).
--fast-build Do not require database to be deterministically
built when using multiple threads. This is faster,
but does introduce variability in minimizer/LCA
pairs. Used with --build and --standard options.
> kraken2
# kraken2
Need to specify input filenames!
Usage: kraken2 [options] <filename(s)>
Options:
--db NAME Name for Kraken 2 DB
(default: none)
--threads NUM Number of threads (default: 1)
--quick Quick operation (use first hit or hits)
--unclassified-out FILENAME
Print unclassified sequences to filename
--classified-out FILENAME
Print classified sequences to filename
--output FILENAME Print output to filename (default: stdout); "-" will
suppress normal output
--confidence FLOAT Confidence score threshold (default: 0.0); must be
in [0, 1].
--minimum-base-quality NUM
Minimum base quality used in classification (def: 0,
only effective with FASTQ input).
--report FILENAME Print a report with aggregrate counts/clade to file
--use-mpa-style With --report, format report output like Kraken 1's
kraken-mpa-report
--report-zero-counts With --report, report counts for ALL taxa, even if
counts are zero
--memory-mapping Avoids loading database into RAM
--paired The filenames provided have paired-end reads
--use-names Print scientific names instead of just taxids
--gzip-compressed Input files are compressed with gzip
--bzip2-compressed Input files are compressed with bzip2
--help Print this message
--version Print version information
If none of the *-compressed flags are specified, and the filename provided
is a regular file, automatic format detection is attempted.