2018 9/5 bbmerge-auto.sh修正
2019 5/14 BBnormコメント修正、パラメータ修正、ヘルプ追加
bbmap.sh2019 6/13 タイトル修正、6/19 其の3追記
2020 1/29 condaインストール追記、メモリ使用量指定、11/6 誤字修正
2023/02/02 追記
BBtoolsはアメリカのJGIが提供している多機能なNGS向けの解析ツール。2014年にオープンソース化されたらしい。論文は現在準備中とある。アライメントのBBmapや、オーバーラップがないペアリードをマージするBBMerge、エラーコレクションしたfastqを出力するBBnormなど特徴的なコマンドを備える。ここでは以下のコマンドを紹介する。
- BBMap - リファンレンスへのアライメント。
- BBMerge - ペアリードのマージ。k-mer配列を付け足し延長することも可能。
- BBnorm - カバレッジのノーマライズ。
- Dedupe - duplicateしたcontigの除去。
其の2。
其の3。
公式ページ
http://jgi.doe.gov/data-and-tools/bbtools/
ユーザーガイド
http://jgi.doe.gov/data-and-tools/bbtools/bb-tools-user-guide/
ダウンロード
https://sourceforge.net/projects/bbmap/
#homebrew
brew install BBtools
#bioconda (link)
conda install -c bioconda -y bbmap
BBtoolsのコマンドは指定がない限りコア数をあるだけ使う。
> bbmap.sh
$ bbmap.sh
BBMap
Written by Brian Bushnell, from Dec. 2010 - present
Last modified December 20, 2017
Description: Fast and accurate splice-aware read aligner.
Please read bbmap/docs/guides/BBMapGuide.txt for more information.
To index: bbmap.sh ref=<reference fasta>
To map: bbmap.sh in=<reads> out=<output sam>
To map without writing an index:
bbmap.sh ref=<reference fasta> in=<reads> out=<output sam> nodisk
in=stdin will accept reads from standard in, and out=stdout will write to
standard out, but file extensions are still needed to specify the format of the
input and output files e.g. in=stdin.fa.gz will read gzipped fasta from
standard in; out=stdout.sam.gz will write gzipped sam.
Indexing Parameters (required when building the index):
nodisk=f Set to true to build index in memory and write nothing
to disk except output.
ref=<file> Specify the reference sequence. Only do this ONCE,
when building the index (unless using 'nodisk').
build=1 If multiple references are indexed in the same directory,
each needs a unique numeric ID (unless using 'nodisk').
k=13 Kmer length, range 8-15. Longer is faster but uses
more memory. Shorter is more sensitive.
If indexing and mapping are done in two steps, K should
be specified each time.
path=<.> Specify the location to write the index, if you don't
want it in the current working directory.
usemodulo=f Throw away ~80% of kmers based on remainder modulo a
number (reduces RAM by 50% and sensitivity slightly).
Should be enabled both when building the index AND
when mapping.
rebuild=f Force a rebuild of the index (ref= should be set).
Input Parameters:
build=1 Designate index to use. Corresponds to the number
specified when building the index.
in=<file> Primary reads input; required parameter.
in2=<file> For paired reads in two files.
interleaved=auto True forces paired/interleaved input; false forces
single-ended mapping. If not specified, interleaved
status will be autodetected from read names.
fastareadlen=500 Break up FASTA reads longer than this. Max is 500 for
BBMap and 6000 for BBMapPacBio. Only works for FASTA
input (use 'maxlen' for FASTQ input). The default for
bbmap.sh is 500, and for mapPacBio.sh is 6000.
unpigz=f Spawn a pigz (parallel gzip) process for faster
decompression than using Java.
Requires pigz to be installed.
touppercase=t (tuc) Convert lowercase letters in reads to upper case
(otherwise they will not match the reference).
Sampling Parameters:
reads=-1 Set to a positive number N to only process the first N
reads (or pairs), then quit. -1 means use all reads.
samplerate=1 Set to a number from 0 to 1 to randomly select that
fraction of reads for mapping. 1 uses all reads.
skipreads=0 Set to a number N to skip the first N reads (or pairs),
then map the rest.
Mapping Parameters:
fast=f This flag is a macro which sets other paramters to run
faster, at reduced sensitivity. Bad for RNA-seq.
slow=f This flag is a macro which sets other paramters to run
slower, at greater sensitivity. 'vslow' is even slower.
maxindel=16000 Don't look for indels longer than this. Lower is faster.
Set to >=100k for RNAseq with long introns like mammals.
strictmaxindel=f When enabled, do not allow indels longer than 'maxindel'.
By default these are not sought, but may be found anyway.
tipsearch=100 Look this far for read-end deletions with anchors
shorter than K, using brute force.
minid=0.76 Approximate minimum alignment identity to look for.
Higher is faster and less sensitive.
minhits=1 Minimum number of seed hits required for candidate sites.
Higher is faster.
local=f Set to true to use local, rather than global, alignments.
This will soft-clip ugly ends of poor alignments.
perfectmode=f Allow only perfect mappings when set to true (very fast).
semiperfectmode=f Allow only perfect and semiperfect (perfect except for
N's in the reference) mappings.
threads=auto (t) Set to number of threads desired. By default, uses
all cores available.
ambiguous=best (ambig) Set behavior on ambiguously-mapped reads (with
multiple top-scoring mapping locations).
best (use the first best site)
toss (consider unmapped)
random (select one top-scoring site randomly)
all (retain all top-scoring sites)
samestrandpairs=f (ssp) Specify whether paired reads should map to the
same strand or opposite strands.
requirecorrectstrand=t (rcs) Forbid pairing of reads without correct strand
orientation. Set to false for long-mate-pair libraries.
killbadpairs=f (kbp) If a read pair is mapped with an inappropriate
insert size or orientation, the read with the lower
mapping quality is marked unmapped.
pairedonly=f (po) Treat unpaired reads as unmapped. Thus they will
be sent to 'outu' but not 'outm'.
rcomp=f Reverse complement both reads prior to mapping (for LMP
outward-facing libraries).
rcompmate=f Reverse complement read2 prior to mapping.
pairlen=32000 Set max allowed distance between paired reads.
(insert size)=(pairlen)+(read1 length)+(read2 length)
rescuedist=1200 Don't try to rescue paired reads if avg. insert size
greater than this. Lower is faster.
rescuemismatches=32 Maximum mismatches allowed in a rescued read. Lower
is faster.
averagepairdist=100 (apd) Initial average distance between paired reads.
Varies dynamically; does not need to be specified.
deterministic=f Run in deterministic mode. In this case it is good
to set averagepairdist. BBMap is deterministic
without this flag if using single-ended reads,
or run singlethreaded.
bandwidthratio=0 (bwr) If above zero, restrict alignment band to this
fraction of read length. Faster but less accurate.
bandwidth=0 (bw) Set the bandwidth directly.
fraction of read length. Faster but less accurate.
usejni=f (jni) Do alignments faster, in C code. Requires
compiling the C code; details are in /jni/README.txt.
maxsites2=800 Don't analyze (or print) more than this many alignments
per read.
ignorefrequentkmers=t (ifk) Discard low-information kmers that occur often.
excludefraction=0.03 (ef) Fraction of kmers to ignore. For example, 0.03
will ignore the most common 3% of kmers.
greedy=t Use a greedy algorithm to discard the least-useful
kmers on a per-read basis.
kfilter=0 If positive, potential mapping sites must have at
least this many consecutive exact matches.
Quality and Trimming Parameters:
qin=auto Set to 33 or 64 to specify input quality value ASCII
offset. 33 is Sanger, 64 is old Solexa.
qout=auto Set to 33 or 64 to specify output quality value ASCII
offset (only if output format is fastq).
qtrim=f Quality-trim ends before mapping. Options are:
'f' (false), 'l' (left), 'r' (right), and 'lr' (both).
untrim=f Undo trimming after mapping. Untrimmed bases will be
soft-clipped in cigar strings.
trimq=6 Trim regions with average quality below this
(phred algorithm).
mintrimlength=60 (mintl) Don't trim reads to be shorter than this.
fakefastaquality=-1 (ffq) Set to a positive number 1-50 to generate fake
quality strings for fasta input reads.
ignorebadquality=f (ibq) Keep going, rather than crashing, if a read has
out-of-range quality values.
usequality=t Use quality scores when determining which read kmers
to use as seeds.
minaveragequality=0 (maq) Do not map reads with average quality below this.
maqb=0 If positive, calculate maq from this many initial bases.
Output Parameters:
out=<file> Write all reads to this file.
outu=<file> Write only unmapped reads to this file. Does not
include unmapped paired reads with a mapped mate.
outm=<file> Write only mapped reads to this file. Includes
unmapped paired reads with a mapped mate.
mappedonly=f If true, treats 'out' like 'outm'.
bamscript=<file> (bs) Write a shell script to <file> that will turn
the sam output into a sorted, indexed bam file.
ordered=f Set to true to output reads in same order as input.
Slower and uses more memory.
overwrite=f (ow) Allow process to overwrite existing files.
secondary=f Print secondary alignments.
sssr=0.95 (secondarysitescoreratio) Print only secondary alignments
with score of at least this fraction of primary.
ssao=f (secondarysiteasambiguousonly) Only print secondary
alignments for ambiguously-mapped reads.
maxsites=5 Maximum number of total alignments to print per read.
Only relevant when secondary=t.
quickmatch=f Generate cigar strings more quickly.
trimreaddescriptions=f (trd) Truncate read and ref names at the first whitespace,
assuming that the remainder is a comment or description.
ziplevel=2 (zl) Compression level for zip or gzip output.
pigz=f Spawn a pigz (parallel gzip) process for faster
compression than Java. Requires pigz to be installed.
machineout=f Set to true to output statistics in machine-friendly
'key=value' format.
printunmappedcount=f Print the total number of unmapped reads and bases.
If input is paired, the number will be of pairs
for which both reads are unmapped.
showprogress=0 If positive, print a '.' every X reads.
showprogress2=0 If positive, print the number of seconds since the
last progress update (instead of a '.').
renamebyinsert=f Renames reads based on their mapped insert size.
Post-Filtering Parameters:
idfilter=0 Independant of minid; sets exact minimum identity
allowed for alignments to be printed. Range 0 to 1.
subfilter=-1 Ban alignments with more than this many substitutions.
insfilter=-1 Ban alignments with more than this many insertions.
delfilter=-1 Ban alignments with more than this many deletions.
indelfilter=-1 Ban alignments with more than this many indels.
editfilter=-1 Ban alignments with more than this many edits.
inslenfilter=-1 Ban alignments with an insertion longer than this.
dellenfilter=-1 Ban alignments with a deletion longer than this.
nfilter=-1 Ban alignments with more than this many ns. This
includes nocall, noref, and off scaffold ends.
Sam flags and settings:
noheader=f Disable generation of header lines.
sam=1.4 Set to 1.4 to write Sam version 1.4 cigar strings,
with = and X, or 1.3 to use M.
saa=t (secondaryalignmentasterisks) Use asterisks instead of
bases for sam secondary alignments.
cigar=t Set to 'f' to skip generation of cigar strings (faster).
keepnames=f Keep original names of paired reads, rather than
ensuring both reads have the same name.
intronlen=999999999 Set to a lower number like 10 to change 'D' to 'N' in
cigar strings for deletions of at least that length.
rgid= Set readgroup ID. All other readgroup fields
can be set similarly, with the flag rgXX=
mdtag=f Write MD tags.
nhtag=f Write NH tags.
xmtag=f Write XM tags (may only work correctly with ambig=all).
amtag=f Write AM tags.
nmtag=f Write NM tags.
xstag=f Set to 'xs=fs', 'xs=ss', or 'xs=us' to write XS tags
for RNAseq using firststrand, secondstrand, or
unstranded libraries. Needed by Cufflinks.
JGI mainly uses 'firststrand'.
stoptag=f Write a tag indicating read stop location, prefixed by YS:i:
lengthtag=f Write a tag indicating (query,ref) alignment lengths,
prefixed by YL:Z:
idtag=f Write a tag indicating percent identity, prefixed by YI:f:
inserttag=f Write a tag indicating insert size, prefixed by X8:Z:
scoretag=f Write a tag indicating BBMap's raw score, prefixed by YR:i:
timetag=f Write a tag indicating this read's mapping time, prefixed by X0:i:
boundstag=f Write a tag indicating whether either read in the pair
goes off the end of the reference, prefixed by XB:Z:
notags=f Turn off all optional tags.
Histogram and statistics output parameters:
scafstats=<file> Statistics on how many reads mapped to which scaffold.
refstats=<file> Statistics on how many reads mapped to which reference
file; only for BBSplit.
sortscafs=t Sort scaffolds or references by read count.
bhist=<file> Base composition histogram by position.
qhist=<file> Quality histogram by position.
aqhist=<file> Histogram of average read quality.
bqhist=<file> Quality histogram designed for box plots.
lhist=<file> Read length histogram.
ihist=<file> Write histogram of insert sizes (for paired reads).
ehist=<file> Errors-per-read histogram.
qahist=<file> Quality accuracy histogram of error rates versus
quality score.
indelhist=<file> Indel length histogram.
mhist=<file> Histogram of match, sub, del, and ins rates by
read location.
gchist=<file> Read GC content histogram.
gcbins=100 Number gchist bins. Set to 'auto' to use read length.
gcpairs=t Use average GC of paired reads.
idhist=<file> Histogram of read count versus percent identity.
idbins=100 Number idhist bins. Set to 'auto' to use read length.
statsfile=stderr Mapping statistics are printed here.
Coverage output parameters (these may reduce speed and use more RAM):
covstats=<file> Per-scaffold coverage info.
rpkm=<file> Per-scaffold RPKM/FPKM counts.
covhist=<file> Histogram of # occurrences of each depth level.
basecov=<file> Coverage per base location.
bincov=<file> Print binned coverage per location (one line per X bases).
covbinsize=1000 Set the binsize for binned coverage output.
nzo=t Only print scaffolds with nonzero coverage.
twocolumn=f Change to true to print only ID and Avg_fold instead of
all 6 columns to the 'out=' file.
32bit=f Set to true if you need per-base coverage over 64k.
strandedcov=f Track coverage for plus and minus strand independently.
startcov=f Only track start positions of reads.
secondarycov=t Include coverage of secondary alignments.
physcov=f Calculate physical coverage for paired reads.
This includes the unsequenced bases.
delcoverage=t (delcov) Count bases covered by deletions as covered.
True is faster than false.
covk=0 If positive, calculate kmer coverage statistics.
Java Parameters:
-Xmx This will be passed to Java to set memory usage,
overriding the program's automatic memory detection.
-Xmx20g will specify 20 gigs of RAM, and -Xmx800m
will specify 800 megs. The max is typically 85% of
physical memory. The human genome requires around 24g,
or 12g with the 'usemodulo' flag. The index uses
roughly 6 bytes per reference base.
-eoom This flag will cause the process to exit if an
out-of-memory exception occurs. Requires Java 8u92+.
-da Disable assertions.
Please contact Brian Bushnell at bbushnell@lbl.gov if you encounter
any problems, or post at: http://seqanswers.com/forums/showthread.php?t=41057
> bbnorm.sh
$ bbnorm.sh
Written by Brian Bushnell
Last modified October 19, 2017
Description: Normalizes read depth based on kmer counts.
Can also error-correct, bin reads by kmer depth, and generate a kmer depth histogram.
However, Tadpole has superior error-correction to BBNorm.
Please read bbmap/docs/guides/BBNormGuide.txt for more information.
Usage: bbnorm.sh in=<input> out=<reads to keep> outt=<reads to toss> hist=<histogram output>
Input parameters:
in=null Primary input. Use in2 for paired reads in a second file
in2=null Second input file for paired reads in two files
extra=null Additional files to use for input (generating hash table) but not for output
fastareadlen=2^31 Break up FASTA reads longer than this. Can be useful when processing scaffolded genomes
tablereads=-1 Use at most this many reads when building the hashtable (-1 means all)
kmersample=1 Process every nth kmer, and skip the rest
readsample=1 Process every nth read, and skip the rest
interleaved=auto May be set to true or false to force the input read file to ovverride autodetection of the input file as paired interleaved.
qin=auto ASCII offset for input quality. May be 33 (Sanger), 64 (Illumina), or auto.
Output parameters:
out=<file> File for normalized or corrected reads. Use out2 for paired reads in a second file
outt=<file> (outtoss) File for reads that were excluded from primary output
reads=-1 Only process this number of reads, then quit (-1 means all)
sampleoutput=t Use sampling on output as well as input (not used if sample rates are 1)
keepall=f Set to true to keep all reads (e.g. if you just want error correction).
zerobin=f Set to true if you want kmers with a count of 0 to go in the 0 bin instead of the 1 bin in histograms.
Default is false, to prevent confusion about how there can be 0-count kmers.
The reason is that based on the 'minq' and 'minprob' settings, some kmers may be excluded from the bloom filter.
tmpdir=/var/folders/f2/r9ydwd5d5vxgfzz5h48grzgc0000gn/T/ This will specify a directory for temp files (only needed for multipass runs). If null, they will be written to the output directory.
usetempdir=t Allows enabling/disabling of temporary directory; if disabled, temp files will be written to the output directory.
qout=auto ASCII offset for output quality. May be 33 (Sanger), 64 (Illumina), or auto (same as input).
rename=f Rename reads based on their kmer depth.
Hashing parameters:
k=31 Kmer length (values under 32 are most efficient, but arbitrarily high values are supported)
bits=32 Bits per cell in bloom filter; must be 2, 4, 8, 16, or 32. Maximum kmer depth recorded is 2^cbits. Automatically reduced to 16 in 2-pass.
Large values decrease accuracy for a fixed amount of memory, so use the lowest number you can that will still capture highest-depth kmers.
hashes=3 Number of times each kmer is hashed and stored. Higher is slower.
Higher is MORE accurate if there is enough memory, and LESS accurate if there is not enough memory.
prefilter=f True is slower, but generally more accurate; filters out low-depth kmers from the main hashtable. The prefilter is more memory-efficient because it uses 2-bit cells.
prehashes=2 Number of hashes for prefilter.
prefilterbits=2 (pbits) Bits per cell in prefilter.
prefiltersize=0.35 Fraction of memory to allocate to prefilter.
buildpasses=1 More passes can sometimes increase accuracy by iteratively removing low-depth kmers
minq=6 Ignore kmers containing bases with quality below this
minprob=0.5 Ignore kmers with overall probability of correctness below this
threads=auto (t) Spawn exactly X hashing threads (default is number of logical processors). Total active threads may exceed X due to I/O threads.
rdk=t (removeduplicatekmers) When true, a kmer's count will only be incremented once per read pair, even if that kmer occurs more than once.
Normalization parameters:
fixspikes=f (fs) Do a slower, high-precision bloom filter lookup of kmers that appear to have an abnormally high depth due to collisions.
target=100 (tgt) Target normalization depth. NOTE: All depth parameters control kmer depth, not read depth.
For kmer depth Dk, read depth Dr, read length R, and kmer size K: Dr=Dk*(R/(R-K+1))
maxdepth=-1 (max) Reads will not be downsampled when below this depth, even if they are above the target depth.
mindepth=5 (min) Kmers with depth below this number will not be included when calculating the depth of a read.
minkmers=15 (mgkpr) Reads must have at least this many kmers over min depth to be retained. Aka 'mingoodkmersperread'.
percentile=54.0 (dp) Read depth is by default inferred from the 54th percentile of kmer depth, but this may be changed to any number 1-100.
uselowerdepth=t (uld) For pairs, use the depth of the lower read as the depth proxy.
deterministic=t (dr) Generate random numbers deterministically to ensure identical output between multiple runs. May decrease speed with a huge number of threads.
passes=2 (p) 1 pass is the basic mode. 2 passes (default) allows greater accuracy, error detection, better contol of output depth.
Error detection parameters:
hdp=90.0 (highdepthpercentile) Position in sorted kmer depth array used as proxy of a read's high kmer depth.
ldp=25.0 (lowdepthpercentile) Position in sorted kmer depth array used as proxy of a read's low kmer depth.
tossbadreads=f (tbr) Throw away reads detected as containing errors.
requirebothbad=f (rbb) Only toss bad pairs if both reads are bad.
errordetectratio=125 (edr) Reads with a ratio of at least this much between their high and low depth kmers will be classified as error reads.
highthresh=12 (ht) Threshold for high kmer. A high kmer at this or above are considered non-error.
lowthresh=3 (lt) Threshold for low kmer. Kmers at this and below are always considered errors.
Error correction parameters:
ecc=f Set to true to correct errors. NOTE: Tadpole is now preferred for ecc as it does a better job.
ecclimit=3 Correct up to this many errors per read. If more are detected, the read will remain unchanged.
errorcorrectratio=140 (ecr) Adjacent kmers with a depth ratio of at least this much between will be classified as an error.
echighthresh=22 (echt) Threshold for high kmer. A kmer at this or above may be considered non-error.
eclowthresh=2 (eclt) Threshold for low kmer. Kmers at this and below are considered errors.
eccmaxqual=127 Do not correct bases with quality above this value.
aec=f (aggressiveErrorCorrection) Sets more aggressive values of ecr=100, ecclimit=7, echt=16, eclt=3.
cec=f (conservativeErrorCorrection) Sets more conservative values of ecr=180, ecclimit=2, echt=30, eclt=1, sl=4, pl=4.
meo=f (markErrorsOnly) Marks errors by reducing quality value of suspected errors; does not correct anything.
mue=t (markUncorrectableErrors) Marks errors only on uncorrectable reads; requires 'ecc=t'.
overlap=f (ecco) Error correct by read overlap.
Depth binning parameters:
lowbindepth=10 (lbd) Cutoff for low depth bin.
highbindepth=80 (hbd) Cutoff for high depth bin.
outlow=<file> Pairs in which both reads have a median below lbd go into this file.
outhigh=<file> Pairs in which both reads have a median above hbd go into this file.
outmid=<file> All other pairs go into this file.
Histogram parameters:
hist=<file> Specify a file to write the input kmer depth histogram.
histout=<file> Specify a file to write the output kmer depth histogram.
histcol=3 (histogramcolumns) Number of histogram columns, 2 or 3.
pzc=f (printzerocoverage) Print lines in the histogram with zero coverage.
histlen=1048576 Max kmer depth displayed in histogram. Also affects statistics displayed, but does not affect normalization.
Peak calling parameters:
peaks=<file> Write the peaks to this file. Default is stdout.
minHeight=2 (h) Ignore peaks shorter than this.
minVolume=2 (v) Ignore peaks with less area than this.
minWidth=2 (w) Ignore peaks narrower than this.
minPeak=2 (minp) Ignore peaks with an X-value below this.
maxPeak=BIG (maxp) Ignore peaks with an X-value above this.
maxPeakCount=8 (maxpc) Print up to this many peaks (prioritizing height).
Java Parameters:
-Xmx This will be passed to Java to set memory usage, overriding the program's automatic memory detection.
-Xmx20g will specify 20 gigs of RAM, and -Xmx200m will specify 200 megs. The max is typically 85% of physical memory.
-eoom This flag will cause the process to exit if an out-of-memory exception occurs. Requires Java 8u92+.
-da Disable assertions.
Please contact Brian Bushnell at bbushnell@lbl.gov if you encounter any problems.
> mapPacBio.sh
$ mapPacBio.sh
BBMap
Written by Brian Bushnell, from Dec. 2010 - present
Last modified December 20, 2017
Description: Fast and accurate splice-aware read aligner.
Please read bbmap/docs/guides/BBMapGuide.txt for more information.
To index: bbmap.sh ref=<reference fasta>
To map: bbmap.sh in=<reads> out=<output sam>
To map without writing an index:
bbmap.sh ref=<reference fasta> in=<reads> out=<output sam> nodisk
in=stdin will accept reads from standard in, and out=stdout will write to
standard out, but file extensions are still needed to specify the format of the
input and output files e.g. in=stdin.fa.gz will read gzipped fasta from
standard in; out=stdout.sam.gz will write gzipped sam.
Indexing Parameters (required when building the index):
nodisk=f Set to true to build index in memory and write nothing
to disk except output.
ref=<file> Specify the reference sequence. Only do this ONCE,
when building the index (unless using 'nodisk').
build=1 If multiple references are indexed in the same directory,
each needs a unique numeric ID (unless using 'nodisk').
k=13 Kmer length, range 8-15. Longer is faster but uses
more memory. Shorter is more sensitive.
If indexing and mapping are done in two steps, K should
be specified each time.
path=<.> Specify the location to write the index, if you don't
want it in the current working directory.
usemodulo=f Throw away ~80% of kmers based on remainder modulo a
number (reduces RAM by 50% and sensitivity slightly).
Should be enabled both when building the index AND
when mapping.
rebuild=f Force a rebuild of the index (ref= should be set).
Input Parameters:
build=1 Designate index to use. Corresponds to the number
specified when building the index.
in=<file> Primary reads input; required parameter.
in2=<file> For paired reads in two files.
interleaved=auto True forces paired/interleaved input; false forces
single-ended mapping. If not specified, interleaved
status will be autodetected from read names.
fastareadlen=500 Break up FASTA reads longer than this. Max is 500 for
BBMap and 6000 for BBMapPacBio. Only works for FASTA
input (use 'maxlen' for FASTQ input). The default for
bbmap.sh is 500, and for mapPacBio.sh is 6000.
unpigz=f Spawn a pigz (parallel gzip) process for faster
decompression than using Java.
Requires pigz to be installed.
touppercase=t (tuc) Convert lowercase letters in reads to upper case
(otherwise they will not match the reference).
Sampling Parameters:
reads=-1 Set to a positive number N to only process the first N
reads (or pairs), then quit. -1 means use all reads.
samplerate=1 Set to a number from 0 to 1 to randomly select that
fraction of reads for mapping. 1 uses all reads.
skipreads=0 Set to a number N to skip the first N reads (or pairs),
then map the rest.
Mapping Parameters:
fast=f This flag is a macro which sets other paramters to run
faster, at reduced sensitivity. Bad for RNA-seq.
slow=f This flag is a macro which sets other paramters to run
slower, at greater sensitivity. 'vslow' is even slower.
maxindel=16000 Don't look for indels longer than this. Lower is faster.
Set to >=100k for RNAseq with long introns like mammals.
strictmaxindel=f When enabled, do not allow indels longer than 'maxindel'.
By default these are not sought, but may be found anyway.
tipsearch=100 Look this far for read-end deletions with anchors
shorter than K, using brute force.
minid=0.76 Approximate minimum alignment identity to look for.
Higher is faster and less sensitive.
minhits=1 Minimum number of seed hits required for candidate sites.
Higher is faster.
local=f Set to true to use local, rather than global, alignments.
This will soft-clip ugly ends of poor alignments.
perfectmode=f Allow only perfect mappings when set to true (very fast).
semiperfectmode=f Allow only perfect and semiperfect (perfect except for
N's in the reference) mappings.
threads=auto (t) Set to number of threads desired. By default, uses
all cores available.
ambiguous=best (ambig) Set behavior on ambiguously-mapped reads (with
multiple top-scoring mapping locations).
best (use the first best site)
toss (consider unmapped)
random (select one top-scoring site randomly)
all (retain all top-scoring sites)
samestrandpairs=f (ssp) Specify whether paired reads should map to the
same strand or opposite strands.
requirecorrectstrand=t (rcs) Forbid pairing of reads without correct strand
orientation. Set to false for long-mate-pair libraries.
killbadpairs=f (kbp) If a read pair is mapped with an inappropriate
insert size or orientation, the read with the lower
mapping quality is marked unmapped.
pairedonly=f (po) Treat unpaired reads as unmapped. Thus they will
be sent to 'outu' but not 'outm'.
rcomp=f Reverse complement both reads prior to mapping (for LMP
outward-facing libraries).
rcompmate=f Reverse complement read2 prior to mapping.
pairlen=32000 Set max allowed distance between paired reads.
(insert size)=(pairlen)+(read1 length)+(read2 length)
rescuedist=1200 Don't try to rescue paired reads if avg. insert size
greater than this. Lower is faster.
rescuemismatches=32 Maximum mismatches allowed in a rescued read. Lower
is faster.
averagepairdist=100 (apd) Initial average distance between paired reads.
Varies dynamically; does not need to be specified.
deterministic=f Run in deterministic mode. In this case it is good
to set averagepairdist. BBMap is deterministic
without this flag if using single-ended reads,
or run singlethreaded.
bandwidthratio=0 (bwr) If above zero, restrict alignment band to this
fraction of read length. Faster but less accurate.
bandwidth=0 (bw) Set the bandwidth directly.
fraction of read length. Faster but less accurate.
usejni=f (jni) Do alignments faster, in C code. Requires
compiling the C code; details are in /jni/README.txt.
maxsites2=800 Don't analyze (or print) more than this many alignments
per read.
ignorefrequentkmers=t (ifk) Discard low-information kmers that occur often.
excludefraction=0.03 (ef) Fraction of kmers to ignore. For example, 0.03
will ignore the most common 3% of kmers.
greedy=t Use a greedy algorithm to discard the least-useful
kmers on a per-read basis.
kfilter=0 If positive, potential mapping sites must have at
least this many consecutive exact matches.
Quality and Trimming Parameters:
qin=auto Set to 33 or 64 to specify input quality value ASCII
offset. 33 is Sanger, 64 is old Solexa.
qout=auto Set to 33 or 64 to specify output quality value ASCII
offset (only if output format is fastq).
qtrim=f Quality-trim ends before mapping. Options are:
'f' (false), 'l' (left), 'r' (right), and 'lr' (both).
untrim=f Undo trimming after mapping. Untrimmed bases will be
soft-clipped in cigar strings.
trimq=6 Trim regions with average quality below this
(phred algorithm).
mintrimlength=60 (mintl) Don't trim reads to be shorter than this.
fakefastaquality=-1 (ffq) Set to a positive number 1-50 to generate fake
quality strings for fasta input reads.
ignorebadquality=f (ibq) Keep going, rather than crashing, if a read has
out-of-range quality values.
usequality=t Use quality scores when determining which read kmers
to use as seeds.
minaveragequality=0 (maq) Do not map reads with average quality below this.
maqb=0 If positive, calculate maq from this many initial bases.
Output Parameters:
out=<file> Write all reads to this file.
outu=<file> Write only unmapped reads to this file. Does not
include unmapped paired reads with a mapped mate.
outm=<file> Write only mapped reads to this file. Includes
unmapped paired reads with a mapped mate.
mappedonly=f If true, treats 'out' like 'outm'.
bamscript=<file> (bs) Write a shell script to <file> that will turn
the sam output into a sorted, indexed bam file.
ordered=f Set to true to output reads in same order as input.
Slower and uses more memory.
overwrite=f (ow) Allow process to overwrite existing files.
secondary=f Print secondary alignments.
sssr=0.95 (secondarysitescoreratio) Print only secondary alignments
with score of at least this fraction of primary.
ssao=f (secondarysiteasambiguousonly) Only print secondary
alignments for ambiguously-mapped reads.
maxsites=5 Maximum number of total alignments to print per read.
Only relevant when secondary=t.
quickmatch=f Generate cigar strings more quickly.
trimreaddescriptions=f (trd) Truncate read and ref names at the first whitespace,
assuming that the remainder is a comment or description.
ziplevel=2 (zl) Compression level for zip or gzip output.
pigz=f Spawn a pigz (parallel gzip) process for faster
compression than Java. Requires pigz to be installed.
machineout=f Set to true to output statistics in machine-friendly
'key=value' format.
printunmappedcount=f Print the total number of unmapped reads and bases.
If input is paired, the number will be of pairs
for which both reads are unmapped.
showprogress=0 If positive, print a '.' every X reads.
showprogress2=0 If positive, print the number of seconds since the
last progress update (instead of a '.').
renamebyinsert=f Renames reads based on their mapped insert size.
Post-Filtering Parameters:
idfilter=0 Independant of minid; sets exact minimum identity
allowed for alignments to be printed. Range 0 to 1.
subfilter=-1 Ban alignments with more than this many substitutions.
insfilter=-1 Ban alignments with more than this many insertions.
delfilter=-1 Ban alignments with more than this many deletions.
indelfilter=-1 Ban alignments with more than this many indels.
editfilter=-1 Ban alignments with more than this many edits.
inslenfilter=-1 Ban alignments with an insertion longer than this.
dellenfilter=-1 Ban alignments with a deletion longer than this.
nfilter=-1 Ban alignments with more than this many ns. This
includes nocall, noref, and off scaffold ends.
Sam flags and settings:
noheader=f Disable generation of header lines.
sam=1.4 Set to 1.4 to write Sam version 1.4 cigar strings,
with = and X, or 1.3 to use M.
saa=t (secondaryalignmentasterisks) Use asterisks instead of
bases for sam secondary alignments.
cigar=t Set to 'f' to skip generation of cigar strings (faster).
keepnames=f Keep original names of paired reads, rather than
ensuring both reads have the same name.
intronlen=999999999 Set to a lower number like 10 to change 'D' to 'N' in
cigar strings for deletions of at least that length.
rgid= Set readgroup ID. All other readgroup fields
can be set similarly, with the flag rgXX=
mdtag=f Write MD tags.
nhtag=f Write NH tags.
xmtag=f Write XM tags (may only work correctly with ambig=all).
amtag=f Write AM tags.
nmtag=f Write NM tags.
xstag=f Set to 'xs=fs', 'xs=ss', or 'xs=us' to write XS tags
for RNAseq using firststrand, secondstrand, or
unstranded libraries. Needed by Cufflinks.
JGI mainly uses 'firststrand'.
stoptag=f Write a tag indicating read stop location, prefixed by YS:i:
lengthtag=f Write a tag indicating (query,ref) alignment lengths,
prefixed by YL:Z:
idtag=f Write a tag indicating percent identity, prefixed by YI:f:
inserttag=f Write a tag indicating insert size, prefixed by X8:Z:
scoretag=f Write a tag indicating BBMap's raw score, prefixed by YR:i:
timetag=f Write a tag indicating this read's mapping time, prefixed by X0:i:
boundstag=f Write a tag indicating whether either read in the pair
goes off the end of the reference, prefixed by XB:Z:
notags=f Turn off all optional tags.
Histogram and statistics output parameters:
scafstats=<file> Statistics on how many reads mapped to which scaffold.
refstats=<file> Statistics on how many reads mapped to which reference
file; only for BBSplit.
sortscafs=t Sort scaffolds or references by read count.
bhist=<file> Base composition histogram by position.
qhist=<file> Quality histogram by position.
aqhist=<file> Histogram of average read quality.
bqhist=<file> Quality histogram designed for box plots.
lhist=<file> Read length histogram.
ihist=<file> Write histogram of insert sizes (for paired reads).
ehist=<file> Errors-per-read histogram.
qahist=<file> Quality accuracy histogram of error rates versus
quality score.
indelhist=<file> Indel length histogram.
mhist=<file> Histogram of match, sub, del, and ins rates by
read location.
gchist=<file> Read GC content histogram.
gcbins=100 Number gchist bins. Set to 'auto' to use read length.
gcpairs=t Use average GC of paired reads.
idhist=<file> Histogram of read count versus percent identity.
idbins=100 Number idhist bins. Set to 'auto' to use read length.
statsfile=stderr Mapping statistics are printed here.
Coverage output parameters (these may reduce speed and use more RAM):
covstats=<file> Per-scaffold coverage info.
rpkm=<file> Per-scaffold RPKM/FPKM counts.
covhist=<file> Histogram of # occurrences of each depth level.
basecov=<file> Coverage per base location.
bincov=<file> Print binned coverage per location (one line per X bases).
covbinsize=1000 Set the binsize for binned coverage output.
nzo=t Only print scaffolds with nonzero coverage.
twocolumn=f Change to true to print only ID and Avg_fold instead of
all 6 columns to the 'out=' file.
32bit=f Set to true if you need per-base coverage over 64k.
strandedcov=f Track coverage for plus and minus strand independently.
startcov=f Only track start positions of reads.
secondarycov=t Include coverage of secondary alignments.
physcov=f Calculate physical coverage for paired reads.
This includes the unsequenced bases.
delcoverage=t (delcov) Count bases covered by deletions as covered.
True is faster than false.
covk=0 If positive, calculate kmer coverage statistics.
Java Parameters:
-Xmx This will be passed to Java to set memory usage,
overriding the program's automatic memory detection.
-Xmx20g will specify 20 gigs of RAM, and -Xmx800m
will specify 800 megs. The max is typically 85% of
physical memory. The human genome requires around 24g,
or 12g with the 'usemodulo' flag. The index uses
roughly 6 bytes per reference base.
-eoom This flag will cause the process to exit if an
out-of-memory exception occurs. Requires Java 8u92+.
-da Disable assertions.
Please contact Brian Bushnell at bbushnell@lbl.gov if you encounter
any problems, or post at: http://seqanswers.com/forums/showthread.php?t=41057
> stats.sh
$ stats.sh
Written by Brian Bushnell
Last modified December 7, 2017
Description: Generates basic assembly statistics such as scaffold count,
N50, L50, GC content, gap percent, etc. For multiple files, please use
statswrapper.sh. Works with fasta and fastq only (gzipped is fine).
Please read bbmap/docs/guides/StatsGuide.txt for more information.
Usage: stats.sh in=<file>
Parameters:
in=file Specify the input fasta file, or stdin.
gc=file Writes ACGTN content per scaffold to a file.
gchist=file Filename to output scaffold gc content histogram.
shist=file Filename to output cumulative scaffold length histogram.
gcbins=200 Number of bins for gc histogram.
n=10 Number of contiguous Ns to signify a break between contigs.
k=13 Estimate memory usage of BBMap with this kmer length.
minscaf=0 Ignore scaffolds shorter than this.
phs=f (printheaderstats) Set to true to print total size of headers.
n90=t (printn90) Print the N/L90 metrics.
extended=f Print additional metrics such as N/L90 and log sum.
logoffset=1000 Minimum length for calculating log sum.
logbase=2 Log base for calculating log sum.
pdl=f (printduplicatelines) Set to true to print lines in the
scaffold size table where the counts did not change.
n_=t This flag will prefix the terms 'contigs' and 'scaffolds'
with 'n_' in formats 3-6.
addname=f Adds a column for input file name, for formats 3-6.
format=<0-7> Format of the stats information; default 1.
format=0 prints no assembly stats.
format=1 uses variable units like MB and KB, and is designed for compatibility with existing tools.
format=2 uses only whole numbers of bases, with no commas in numbers, and is designed for machine parsing.
format=3 outputs stats in 2 rows of tab-delimited columns: a header row and a data row.
format=4 is like 3 but with scaffold data only.
format=5 is like 3 but with contig data only.
format=6 is like 3 but the header starts with a #.
format=7 is like 1 but only prints contig info.
gcformat=<0-4> Select GC output format; default 1.
gcformat=0: (no base content info printed)
gcformat=1: name length A C G T N GC
gcformat=2: name GC
gcformat=4: name length GC
Note that in gcformat 1, A+C+G+T=1 even when N is nonzero.
Please contact Brian Bushnell at bbushnell@lbl.gov if you encounter any problems.
> bbmerge.sh
$ bbmerge.sh
Written by Brian Bushnell and Jonathan Rood
Last modified March 13, 2017
Description: Merges paired reads into single reads by overlap detection.
With sufficient coverage, can also merge nonoverlapping reads by kmer extension.
Kmer modes requires much more memory, and should be used with the bbmerge-auto.sh script.
Please read bbmap/docs/guides/BBMergeGuide.txt for more information.
Usage for interleaved files: bbmerge.sh in=<reads> out=<merged reads> outu=<unmerged reads>
Usage for paired files: bbmerge.sh in1=<read1> in2=<read2> out=<merged reads> outu1=<unmerged1> outu2=<unmerged2>
Input may be stdin or a file, fasta or fastq, raw or gzipped.
Input parameters:
in=null Primary input. 'in2' will specify a second file.
interleaved=auto May be set to true or false to override autodetection of
whether the input file as interleaved.
reads=-1 Quit after this many read pairs (-1 means all).
Output parameters:
out=<file> File for merged reads. 'out2' will specify a second file.
outu=<file> File for unmerged reads. 'outu2' will specify a second file.
outinsert=<file> (outi) File to write read names and insert sizes.
outadapter=<file> (outa) File to write consensus adapter sequences.
outc=<file> File to write input read kmer cardinality estimate.
ihist=<file> (hist) Insert length histogram output file.
nzo=t Only print histogram bins with nonzero values.
showhiststats=t Print extra header lines with statistical information.
ziplevel=2 Set to 1 (lowest) through 9 (max) to change compression
level; lower compression is faster.
ordered=f Output reads in same order as input.
mix=f Output both the merged (or mergable) and unmerged reads
in the same file (out=). Useful for ecco mode.
Trimming/Filtering parameters:
qtrim=f Trim read ends to remove bases with quality below minq.
Trims BEFORE merging.
Values: t (trim both ends),
f (neither end),
r (right end only),
l (left end only).
qtrim2=f May be specified instead of qtrim to perform trimming
only if merging is unsuccessful, then retry merging.
trimq=10 Trim quality threshold. This may be a comma-delimited
list (ascending) to try multiple values.
minlength=1 (ml) Reads shorter than this after trimming, but before
merging, will be discarded. Pairs will be discarded only
if both are shorter.
maxlength=-1 Reads with longer insert sizes will be discarded.
tbo=f (trimbyoverlap) Trim overlapping reads to remove
rightmost (3') non-overlapping portion, instead of joining.
minavgquality=0 (maq) Reads with average quality below this, after
trimming, will not be attempted to be merged.
maxexpectederrors=0 (mee) If positive, reads with more combined expected
errors than this will not be attempted to be merged.
forcetrimleft=0 (ftl) If nonzero, trim left bases of the read to
this position (exclusive, 0-based).
forcetrimright=0 (ftr) If nonzero, trim right bases of the read
after this position (exclusive, 0-based).
forcetrimright2=0 (ftr2) If positive, trim this many bases on the right end.
forcetrimmod=5 (ftm) If positive, trim length to be equal to
zero modulo this number.
ooi=f Output only incorrectly merged reads, for testing.
trimpolya=t Trim trailing poly-A tail from adapter output. Only
affects outadapter. This also trims poly-A followed
by poly-G, which occurs on NextSeq.
Processing Parameters:
usejni=f (jni) Do overlapping in C code, which is faster. Requires
compiling the C code; details are in /jni/README.txt.
merge=t Create merged reads. If set to false, you can still
generate an insert histogram.
ecco=f Error-correct the overlapping part, but don't merge.
trimnonoverlapping=f (tno) Trim all non-overlapping portions, leaving only
consensus sequence. By default, only sequence to the
right of the overlap (adapter sequence) is trimmed.
useoverlap=t Attempt find the insert size using read overlap.
mininsert=35 Minimum insert size to merge reads.
mininsert0=35 Insert sizes less than this will not be considered.
Must be less than or equal to mininsert.
minoverlap=12 Minimum number of overlapping bases to allow merging.
minoverlap0=8 Overlaps shorter than this will not be considered.
Must be less than or equal to minoverlap.
minq=9 Ignore bases with quality below this.
maxq=41 Cap output quality scores at this.
entropy=t Increase the minimum overlap requirement for low-
complexity reads.
efilter=6 Ban overlaps with over this many times the expected
number of errors. Lower is more strict. -1 disables.
pfilter=0.00004 Ban improbable overlaps. Higher is more strict. 0 will
disable the filter; 1 will allow only perfect overlaps.
kfilter=0 Ban overlaps that create kmers with count below
this value (0 disables). If this is used minprob should
probably be set to 0. Requires good coverage.
ouq=f Calculate best overlap using quality values.
owq=t Calculate best overlap without using quality values.
usequality=t If disabled, quality values are completely ignored,
both for overlap detection and filtering. May be useful
for data with inaccurate quality values.
iupacton=f (itn) Change ambiguous IUPAC symbols to N.
adapter= Specify the adapter sequences used for these reads, if
known; this can be a fasta file or a literal sequence.
Read 1 and 2 can have adapters specified independently
with the adapter1 and adapter2 flags. adapter=default
will use a list of common adapter sequences.
Ratio Mode:
ratiomode=t Score overlaps based on the ratio of matching to
mismatching bases.
maxratio=0.09 Max error rate; higher increases merge rate.
ratiomargin=5.5 Lower increases merge rate; min is 1.
ratiooffset=0.55 Lower increases merge rate; min is 0.
ratiominoverlapreduction=3 This is the difference between minoverlap in
flat mode and minoverlap in ratio mode; generally,
minoverlap should be lower in ratio mode.
minsecondratio=0.1 Cutoff for second-best overlap ratio.
Flat Mode:
flatmode=f Score overlaps based on the total number of mismatching
bases only.
margin=2 The best overlap must have at least 'margin' fewer
mismatches than the second best.
mismatches=3 Do not allow more than this many mismatches.
requireratiomatch=f (rrm) Require the answer from flat mode and ratio mode
to agree, reducing false positives if both are enabled.
trimonfailure=t (tof) If detecting insert size by overlap fails,
the reads will be trimmed and this will be re-attempted.
*** Ratio Mode and Flat Mode may be used alone or simultaneously. ***
*** Ratio Mode is usually more accurate and is the default mode. ***
Strictness (these are mutually exclusive macros that set other parameters):
strict=f Decrease false positive rate and merging rate.
verystrict=f (vstrict) Greatly decrease FP and merging rate.
ultrastrict=f (ustrict) Decrease FP and merging rate even more.
maxstrict=f (xstrict) Maximally decrease FP and merging rate.
loose=f Increase false positive rate and merging rate.
veryloose=f (vloose) Greatly increase FP and merging rate.
ultraloose=f (uloose) Increase FP and merging rate even more.
maxloose=f (xloose) Maximally decrease FP and merging rate.
fast=f Fastest possible mode; less accurate.
Tadpole Parameters (for read extension and error-correction):
k=31 Kmer length. 31 (or less) is fastest and uses the least
memory, but higher values may be more accurate.
60 tends to work well for 150bp reads.
extend=0 Extend reads to the right this much before merging.
Requires sufficient (>5x) kmer coverage.
extend2=0 Extend reads this much only after a failed merge attempt,
or in rem/rsem mode.
iterations=1 (ei) Iteratively attempt to extend by extend2 distance
and merge up to this many times.
rem=f (requireextensionmatch) Do not merge if the predicted
insert size differs before and after extension.
However, if only the extended reads overlap, then that
insert will be used. Requires setting extend2.
rsem=f (requirestrictextensionmatch) Similar to rem but stricter.
Reads will only merge if the predicted insert size before
and after extension match. Requires setting extend2.
ecctadpole=f (ecct) If reads fail to merge, error-correct with Tadpole
and try again. This happens prior to extend2.
reassemble=t If ecct is enabled, use Tadpole's reassemble mode for
error correction. Alternatives are pincer and tail.
removedeadends (shave) Remove kmers leading to dead ends.
removebubbles (rinse) Remove kmers in error bubbles.
mindepthseed=3 (mds) Minimum kmer depth to begin extension.
mindepthextend=2 (mde) Minimum kmer depth continue extension.
branchmult1=20 Min ratio of 1st to 2nd-greatest path depth at high depth.
branchmult2=3 Min ratio of 1st to 2nd-greatest path depth at low depth.
branchlower=3 Max value of 2nd-greatest path depth to be considered low.
ibb=t Ignore backward branches when extending.
extra=<file> A file or comma-delimited list of files of reads to use
for kmer counting, but not for merging or output.
prealloc=f Pre-allocate memory rather than dynamically growing;
faster and more memory-efficient for large datasets.
A float fraction (0-1) may be specified, default 1.
prefilter=0 If set to a positive integer, use a countmin sketch to
ignore kmers with depth of that value or lower, to
reduce memory usage.
minprob=0.5 Ignore kmers with overall probability of correctness
below this, to reduce memory usage.
minapproxoverlap=26 For rem mode, do not merge reads if the extended reads
indicate that the raw reads should have overlapped by
at least this much, but no overlap was found.
Java Parameters:
-Xmx This will be passed to Java to set memory usage,
overriding the program's automatic memory detection.
For example, -Xmx400m will specify 400 MB RAM.
-eoom This flag will cause the process to exit if an
out-of-memory exception occurs. Requires Java 8u92+.
-da Disable assertions.
Please contact Brian Bushnell at bbushnell@lbl.gov if you encounter any problems.
> dedupe.sh
$ dedupe.sh
Written by Brian Bushnell and Jonathan Rood
Last modified November 20, 2017
Description: Accepts one or more files containing sets of sequences (reads or scaffolds).
Removes duplicate sequences, which may be specified to be exact matches, subsequences, or sequences within some percent identity.
Can also find overlapping sequences and group them into clusters.
Please read bbmap/docs/guides/DedupeGuide.txt for more information.
Usage: dedupe.sh in=<file or stdin> out=<file or stdout>
An example of running Dedupe for clustering short reads:
dedupe.sh in=x.fq am=f ac=f fo c pc rnc=f mcs=4 mo=100 s=1 pto cc qin=33 csf=stats.txt pattern=cluster_%.fq dot=graph.dot
Input may be fasta or fastq, compressed or uncompressed.
Output may be stdout or a file. With no output parameter, data will be written to stdout.
If 'out=null', there will be no output, but statistics will still be printed.
You can also use 'dedupe <infile> <outfile>' without the 'in=' and 'out='.
I/O parameters:
in=<file,file> A single file or a comma-delimited list of files.
out=<file> Destination for all output contigs.
pattern=<file> Clusters will be written to individual files, where the '%' symbol in the pattern is replaced by cluster number.
outd=<file> Optional; removed duplicates will go here.
csf=<file> (clusterstatsfile) Write a list of cluster names and sizes.
dot=<file> (graph) Write a graph in dot format. Requires 'fo' and 'pc' flags.
threads=auto (t) Set number of threads to use; default is number of logical processors.
overwrite=t (ow) Set to false to force the program to abort rather than overwrite an existing file.
showspeed=t (ss) Set to 'f' to suppress display of processing speed.
minscaf=0 (ms) Ignore contigs/scaffolds shorter than this.
interleaved=auto If true, forces fastq input to be paired and interleaved.
ziplevel=2 Set to 1 (lowest) through 9 (max) to change compression level; lower compression is faster.
Output format parameters:
storename=t (sn) Store scaffold names (set false to save memory).
#addpairnum=f Add .1 and .2 to numeric id of read1 and read2.
storequality=t (sq) Store quality values for fastq assemblies (set false to save memory).
uniquenames=t (un) Ensure all output scaffolds have unique names. Uses more memory.
numbergraphnodes=t (ngn) Label dot graph nodes with read numbers rather than read names.
sort=f Sort output (otherwise it will be random). Options:
length: Sort by length
quality: Sort by quality
name: Sort by name
id: Sort by input order
ascending=f Sort in ascending order.
ordered=f Output sequences in input order. Equivalent to sort=id ascending.
renameclusters=f (rnc) Rename contigs to indicate which cluster they are in.
printlengthinedges=f (ple) Print the length of contigs in edges.
Processing parameters:
absorbrc=t (arc) Absorb reverse-complements as well as normal orientation.
absorbmatch=t (am) Absorb exact matches of contigs.
absorbcontainment=t (ac) Absorb full containments of contigs.
#absorboverlap=f (ao) Absorb (merge) non-contained overlaps of contigs (TODO).
findoverlap=f (fo) Find overlaps between contigs (containments and non-containments). Necessary for clustering.
uniqueonly=f (uo) If true, all copies of duplicate reads will be discarded, rather than keeping 1.
rmn=f (requirematchingnames) If true, both names and sequence must match.
usejni=f (jni) Do alignments in C code, which is faster, if an edit distance is allowed.
This will require compiling the C code; details are in /jni/README.txt.
Subset parameters:
subsetcount=1 (sstc) Number of subsets used to process the data; higher uses less memory.
subset=0 (sst) Only process reads whose ((ID%subsetcount)==subset).
Clustering parameters:
cluster=f (c) Group overlapping contigs into clusters.
pto=f (preventtransitiveoverlaps) Do not look for new edges between nodes in the same cluster.
minclustersize=1 (mcs) Do not output clusters smaller than this.
pbr=f (pickbestrepresentative) Only output the single highest-quality read per cluster.
Cluster postprocessing parameters:
processclusters=f (pc) Run the cluster processing phase, which performs the selected operations in this category.
For example, pc AND cc must be enabled to perform cc.
fixmultijoins=t (fmj) Remove redundant overlaps between the same two contigs.
removecycles=t (rc) Remove all cycles so clusters form trees.
cc=t (canonicizeclusters) Flip contigs so clusters have a single orientation.
fcc=f (fixcanoncontradictions) Truncate graph at nodes with canonization disputes.
foc=f (fixoffsetcontradictions) Truncate graph at nodes with offset disputes.
mst=f (maxspanningtree) Remove cyclic edges, leaving only the longest edges that form a tree.
Overlap Detection Parameters
exact=t (ex) Only allow exact symbol matches. When false, an 'N' will match any symbol.
touppercase=t (tuc) Convert input bases to upper-case; otherwise, lower-case will not match.
maxsubs=0 (s) Allow up to this many mismatches (substitutions only, no indels). May be set higher than maxedits.
maxedits=0 (e) Allow up to this many edits (subs or indels). Higher is slower.
minidentity=100 (mid) Absorb contained sequences with percent identity of at least this (includes indels).
minlengthpercent=0 (mlp) Smaller contig must be at least this percent of larger contig's length to be absorbed.
minoverlappercent=0 (mop) Overlap must be at least this percent of smaller contig's length to cluster and merge.
minoverlap=200 (mo) Overlap must be at least this long to cluster and merge.
depthratio=0 (dr) When non-zero, overlaps will only be formed between reads with a depth ratio of at most this.
Should be above 1. Depth is determined by parsing the read names; this information can be added
by running KmerNormalize (khist.sh, bbnorm.sh, or ecc.sh) with the flag 'rename'
k=31 Seed length used for finding containments and overlaps. Anything shorter than k will not be found.
numaffixmaps=1 (nam) Number of prefixes/suffixes to index per contig. Higher is more sensitive, if edits are allowed.
hashns=f Set to true to search for matches using kmers containing Ns. Can lead to extreme slowdown in some cases.
#ignoreaffix1=f (ia1) Ignore first affix (for testing).
#storesuffix=f (ss) Store suffix as well as prefix. Automatically set to true when doing inexact matches.
Other Parameters
qtrim=f Set to qtrim=rl to trim leading and trailing Ns.
trimq=6 Quality trim level.
forcetrimleft=-1 (ftl) If positive, trim bases to the left of this position (exclusive, 0-based).
forcetrimright=-1 (ftr) If positive, trim bases to the right of this position (exclusive, 0-based).
Note on Proteins / Amino Acids
Dedupe supports amino acid space via the 'amino' flag. This also changes the default kmer length to 10.
In amino acid mode, all flags related to canonicity and reverse-complementation are disabled,
and nam (numaffixmaps) is currently limited to 2 per tip.
Java Parameters:
-Xmx This will be passed to Java to set memory usage, overriding the program's automatic memory detection.
-Xmx20g will specify 20 gigs of RAM, and -Xmx200m will specify 200 megs. The max is typically 85% of physical memory.
-eoom This flag will cause the process to exit if an out-of-memory exception occurs. Requires Java 8u92+.
-da Disable assertions.
Please contact Brian Bushnell at bbushnell@lbl.gov if you encounter any problems.
ラン
BBMap (リンク)-リードのリファンレンスへのアライメント
BBMapは、k-merに分解してk-merサイズのseedでアライメントと伸長を行うアライナー(デファルトではk=13)。 ショートリードのアライメントだけでなく、ロングリードのアライメントも可能とされ、Illumina,、454、Sanger、Ion Torrent、Pac Bio、Nanoporeのリードのアライメント対応がマニュアルに記載されている。
・index作成とペアリードのアライメント(index=>マッピング=>アライメント)。メモリは最大20GB指定。
bbmap.sh -Xmx20g in1=reads1.fq in2=reads2.fq out=mapped.sam ref=reference.fa
- -Xmx This will be passed to Java to set memory usage, overriding the program's automatic memory detection. -Xmx20g will specify 20 gigs of RAM, and -Xmx800m will specify 800 megs. The max is typically 85% of physical memory. The human genome requires around 24g, or 12g with the 'usemodulo' flag. The index uses roughly 6 bytes per reference base.
indexディレクトリが作られ、リードがリファレンスにアライメントされる。defaultだとマシンの全コアが計算に使用される(明示するなら"t=")。
bbmap.shは700bp以下のリードしか扱えない。pacbioのアライメントは6-kbまでのリードに対応した専用コマンドを使う。
・ロングリードのアライメント。
mapPacBio.sh in=reads.fq out=mapped.sam ref=reference.fa
#リファレンスへのマッピング率(coverage), GCなどの統計データを出力。メモリ100GB指定
mapPacBio.sh in=reads.fq out=mapped.sam ref=reference.fa covstats=mapping_stats.txt nodisk=true -Xmx100g
- nodisk=f Set to true to build index in memory and write nothing to disk except output
6-kb以上のリードは自動で断片化されリネームされてアライメントされる。ナノポアのデータもこのコマンドを使うことが推奨されている。
様々な統計データを出力する。
bbmap.sh in1=reads1.fq in2=reads2.fq bhist=bhist.txt qhist=qhist.txt aqhist=aqhist.txt lhist=lhist.txt ihist=ihist.txt ehist=ehist.txt qahist=qahist.txt indelhist=indelhist.txt mhist=mhist.txt gchist=gchist.txt idhist=idhist.txt scafstats=scafstats.txt
- covstats=<file> Per-scaffold coverage info.
- rpkm=<file> Per-scaffold RPKM/FPKM counts.
- covhist=<file> Histogram of # occurrences of each depth level.
- basecov=<file> Coverage per base location.
- bincov=<file> Print binned coverage per location (one line per X bases).
- covbinsize=1000 Set the binsize for binned co
標準出力には以下のようなデータがプリントされる。
------------------ Results ------------------
Genome: 1
Key Length: 13
Max Indel: 16000
Minimum Score Ratio: 0.56
Mapping Mode: normal
Reads Used: 2647748 (397162200 bases)
Mapping: 70.725 seconds.
Reads/sec: 37437.34
kBases/sec: 5615.60
Pairing data: pct reads num reads pct bases num bases
mated pairs: 99.9427% 1323116 99.9427% 396934800
bad pairs: 0.0178% 236 0.0178% 70800
insert size avg: 601.02
insert 25th %: 566.00
insert median: 600.00
insert 75th %: 634.00
insert std dev: 52.83
insert mode: 602
Read 1 data: pct reads num reads pct bases num bases
mapped: 99.9677% 1323447 99.9677% 198517050
unambiguous: 98.0062% 1297479 98.0062% 194621850
ambiguous: 1.9615% 25968 1.9615% 3895200
low-Q discards: 0.0000% 0 0.0000% 0
perfect best site: 38.6619% 511835 38.6619% 76775250
semiperfect site: 38.6630% 511849 38.6630% 76777350
rescued: 1.2452% 16485
Match Rate: NA NA 99.3118% 197215279
Error Rate: 61.3256% 811612 0.6882% 1366602
Sub Rate: 60.9806% 807046 0.6527% 1296069
Del Rate: 0.5644% 7469 0.0326% 64831
Ins Rate: 0.3730% 4937 0.0029% 5702
N Rate: 0.0000% 0 0.0000% 0
Read 2 data: pct reads num reads pct bases num bases
mapped: 99.9702% 1323479 99.9702% 198521850
unambiguous: 97.9981% 1297371 97.9981% 194605650
ambiguous: 1.9721% 26108 1.9721% 3916200
low-Q discards: 0.0000% 0 0.0000% 0
perfect best site: 38.6839% 512126 38.6839% 76818900
semiperfect site: 38.6856% 512148 38.6856% 76822200
rescued: 1.1880% 15728
Match Rate: NA NA 99.3145% 197221255
Error Rate: 61.3046% 811353 0.6855% 1361264
Sub Rate: 60.9506% 806669 0.6523% 1295263
Del Rate: 0.5679% 7516 0.0306% 60669
Ins Rate: 0.3843% 5086 0.0027% 5332
N Rate: 0.0000% 0 0.0000% 0
Total time: 74.777 seconds.
さらに指定した分析データ(ヒストグラム)が出力される。

ゲノムアセンブリの各contigについて、GC含量、平均リードデプス、マッピングカバー率などの基礎的な統計を出力。
bbmap.sh ref=scaffolds.ref.fa nodisk in1=R1.fq in2=R2.fq covstats=mapping.stats threads=20
mapping.stats
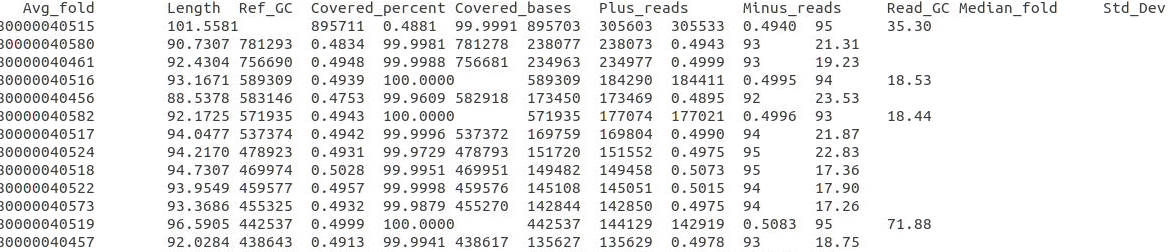
参考1
index作成時のメモリ使用量はリファンレス1塩基につき6 byteとされている。ヒトゲノムでは24GB必要(usemodulo flagをつけて12GB)。
・defaultのk=13の時のメモリ使用量は以下のコマンドで確認できる。
stats.sh in=reference.fa k=13
user$ stats.sh in=../testgenome.fasta k=13
A C G T N IUPAC Other GC GC_stdev
0.2629 0.2364 0.2372 0.2635 0.0000 0.0000 0.0000 0.4736 0.0241
Main genome scaffold total: 8
Main genome contig total: 8
Main genome scaffold sequence total: 3.957 MB
Main genome contig sequence total: 3.957 MB 0.000% gap
Main genome scaffold N/L50: 1/3.573 MB
Main genome contig N/L50: 1/3.573 MB
Main genome scaffold N/L90: 1/3.573 MB
Main genome contig N/L90: 1/3.573 MB
Max scaffold length: 3.573 MB
Max contig length: 3.573 MB
Number of scaffolds > 50 KB: 4
% main genome in scaffolds > 50 KB: 98.63%
Minimum Number Number Total Total Scaffold
Scaffold of of Scaffold Contig Contig
Length Scaffolds Contigs Length Length Coverage
-------- -------------- -------------- -------------- -------------- --------
All 8 8 3,956,956 3,956,956 100.00%
1 KB 8 8 3,956,956 3,956,956 100.00%
2.5 KB 6 6 3,952,233 3,952,233 100.00%
5 KB 6 6 3,952,233 3,952,233 100.00%
10 KB 5 5 3,947,019 3,947,019 100.00%
25 KB 5 5 3,947,019 3,947,019 100.00%
50 KB 4 4 3,902,676 3,902,676 100.00%
100 KB 4 4 3,902,676 3,902,676 100.00%
250 KB 1 1 3,573,470 3,573,470 100.00%
500 KB 1 1 3,573,470 3,573,470 100.00%
1 MB 1 1 3,573,470 3,573,470 100.00%
2.5 MB 1 1 3,573,470 3,573,470 100.00%
BBMap minimum memory estimate at k=13: -Xmx890m (at least 1000 MB physical RAM)
使用したバクテリアゲノムだと1GBの物理メモリが必要と算出された。
参考2
indel部位のスプリットアライメントに対応している。条件はmaxindelフラッグで規定されており、デフォルトではmaxindel=16000となっている。RNA seqでlarge intronが想定される場合などはmaxindel=100000などの大きな数値に指定することが推奨されている。
BBMerge (リンク)-ペアリードのマージ。
BBMegeはオーバーラップするペアエンドリードをマージして1つの長いリードを作るツール。
bbmerge.sh in1=reads1.fq in2=reads2.fq out=merged.fq outu1=unmerged1.fq outu2=unmerged2.fq ihist=ihist.txt
- in=null Primary input. 'in2' will specify a second file.
- out=<file> File for merged reads. 'out2' will specify a second file.
- outu=<file> File for unmerged reads. 'outu2' will specify a second file.
- ihist=<file> (hist) Insert length histogram output file.
インサートサイズのヒストグラムがihist.txtに出力される。マージを厳密に行うならpfilter=1をつける(“pfilter=1” is the strictest possible setting, which bans merges in which there are any mismatches in the overlap region. )。または、verystrict(Greatly decrease FP and merging rate.)をつける。オーサーはアセンブルに使うデータならverystrictをつけ、とにかくマージするならloose(Increase false positive rate and merging rate)をつけることを提案している。
・オーバーラップするペアエンドリードのエラー修復
bbmerge.sh in=interlace.fq out=corrected.fq ecco mix
- mix=f Output both the merged (or mergable) and unmerged reads in the same file (out=). Useful for ecco mode.
- ecco=f Error-correct the overlapping part, but don't merge.
ここでは入力するペアリードはインターレースとなっている。このコマンドを実行することでオーバーラップする領域で塩基が合致する部位はクオリティスコアが上昇し、オーバーラップする領域で塩基が合致しない領域はクオリティスコアが高い方の塩基にbase callが変化したfastqが出力される。マージしたfastq自体は出力されない。クオリティスコアが同じで塩基が合致しない部位はNになる。
・k-merのgraphを利用し、マージされなかったfastqを延長しマージを試みる。
bbmerge-auto.sh in1=reads1.fq in2=reads2.fq out=merged.fq outu=unmerged.fq ihist=ihist.txt ecct extend2=20 iterations=5
- extend2=0 Extend reads this much only after a failed merge attempt, or in rem/rsem mode.
- iterations=1 (ei) Iteratively attempt to extend by extend2 distance and merge up to this many times.
上記のコマンドだと最初にマージが試みられ、ダメならエラー修復して再チャレンジし、それでもダメならk-merのグラフから20-bpくっつけて再マージを試みる。そうして20-bpのextensionを最大5回繰り返す。途中でk-merのgraphがdead endになったり、分岐があったりする試行は中止される。300-bpx2のペアリードでテストすると、最大794-bpのシングルリードが出力された。
・リード長よりインサートサイズが短いペアエンドリードからアダプター配列を見つける。
bbmerge.sh in1=reads1.fq in2=reads2.fq outa=adapters.fa
例えば見つかったアダプターがGATCGGAAGAGCACACGTCTGAACTCCAGTC とする。このアダプター配列を次のコマンドで除く。
bbmerge.sh in1=reads1.fq in2=read2.fq out=merged.fq adapter1=GATCGGAAGAGCACACGTCTGAACTCCAGTC
リードに十分なデプスがあり、アダプター配列も除かれていれば、mergeコマンドは正しく機能する。より長いリードができればアセンブルでより長いk-merを選択することができることになる。一方で、誤ったgraphをつないでしまう可能性もあり、そうなるとアーティファクトの配列ができてしまうことになる(特にレアなindel変異の部位)。
カバレッジがディープな領域をノーマライズして軽量化し、データ解析を行いやすくするツール。例えば、カバレッジが非常に多く変動するシングルセルやウィルスのシーケンスデータ、またメタゲノムデータなどに利用できる。また、シーケンスデータ軽量化のためのdigital normalizationに使える(リンク 真ん中付近)。リードが多すぎて計算リソースを超えている時などに利用する。
bbnorm.sh target=100 min=5 in1=reads1.fq in2=reads2.fq \
out=normalized_1.fq out2=normalized_2.fq
#シングルエンド, k31,mindepth2
bbnorm.sh target=100 min=2 k=31 in1=reads.fq.gz out=normalized.fq.gz
- in=<file> (in1) Input file, or comma-delimited list of files.
- in2=<file> Optional second file for paired reads.
- k=31 Kmer length (values under 32 are most efficient, but arbitrarily high values are supported)
- target=100 (tgt) Target normalization depth. NOTE: All depth parameters control kmer depth, not read depth.
- mindepth=5 (min) Kmers with depth below this number will not be included when calculating the depth of a read.
- maxdepth=-1 (max) Reads will not be downsampled when below this depth, even if they are above the target depth.
- minkmers=15 (mgkpr) Reads must have at least this many kmers over min depth to be retained. Aka 'mingoodkmersperread'.
- out=<file> File for normalized or corrected reads. Use out2 for paired reads in a second file
上記のコマンドではk-merデプスが5以下のリード(シーケンスエラー、またはレアな変異)は排除される。またk-merカバレッジが100になるようにリード数が調整される。
追記;シークエンシングクオリティが高ければロングリードでも使用できる。エラー率が高いリードに適用すると、k-mer一致がないリードが多数出てしまい、多くのリードが失われてしまう可能性が高い。それでも使いたいなら、先にエラーコレクションするか(微細な配列の違いも失われる可能性がある)、k値を少しだけ小さくするかmin=0などにして低い側のカットオフは無くす。
・エラーコレクション
ecc.sh in1=reads1.fq in2=reads2.fq out=corrected.fq ecc=t keepall passes=1 bits=16 prefilter
- ecc=f Set to true to correct errors.
- keepall=f Set to true to keep all reads (e.g. if you just want error correction).
- passes=2 (p) 1 pass is the basic mode. 2 passes (default) allows greater accuracy, error detection, better contol of output depth.
- bits=32 Bits per cell in bloom filter; must be 2, 4, 8, 16, or 32. Maximum kmer depth recorded is 2^cbits. Automatically reduced to 16 in 2-pass.
- prefilter=f True is slower, but generally more accurate; filters out low-depth kmers from the main hashtable. The prefilter is more memory-efficient because it uses 2-bit cells.
エラーコレクション後のfastqがcorrected.fq として出力される。レアk-merのリードのトリミングや除去は一切行われない。
・k-merヒストグラム作成
khist.sh in=reads.fq khist=khist.txt peaks=peaks.txt
ヒストグラムデータの他に、k-mer頻度分布のピークなどをプリントしたサマリーファイルが出力される。
user$ cat peaks.txt
#k 31
#unique_kmers 32597468
#main_peak 14
#genome_size 6316924
#haploid_genome_size 6316924
#fold_coverage 14
#haploid_fold_coverage 14
#ploidy 1
#percent_repeat 36.469
#start center stop max volume
6 14 35 338679 4013227
41 51 121 3023 69382
823 1556 1569 10 1546
1569 1614 1624 21 605
1624 1637 1667 12 455
1667 1683 1705 9 340
1705 1730 1748 13 388
1748 1768 2257 3 1902
3198 4299 4410 4 3095
4410 4501 4523 2 288
4523 4542 5077 2 1112
注意点として、BBnormは発現解析(RNA seqやchip-seq)やレアバリアントの探索などに使ってはならない。 使うとひどいbiasが生じる可能性がある。そのようなデータセットでデータを削減する必要があるなら、ノーマライズでなくランダムサンプリングするツールを使う。
fastqのランダムサンプリングツールとしてseqtkやseqkitを紹介しています。
Dedupe(リンク)-duplicateしたcontigの除去。
k-merベースのde brujin graphでは重複したcontigはほぼ作られないが、overlap layput方式のアセンブルだとduplication contigができることがある。そのようなcontigから重複したcontigを除けるように本コマンドは設計された。また、NCBIのrefseqなどは重複した配列が大量に存在し、blast解析すると同じような配列が大量にhitすることがあるが、そのようなデータベースのキュレーションにも使えるとされる。
また、NCBIの
・duplication sequencesの除去。
dedupe.sh in=X.fa out=Z.fa outd=duplicates.fa
- outd
outdの指定でduplication readsも別出力可能。
・5ミスマッチと2editsまで許容してduplication sequencesを除去。
dedupe.sh in=X.fa out=Y.fa s=5 e=2
s=5だけ指定すると塩基のミスマッチだけ5つまで許容してduplication sequencesが探索される。e=2だけ指定すると、どのような変異でも2つまで許容してduplication sequencesが探索される(例えば2塩基ミスマッチ or 1挿入と1塩基ミスマッチ)。
・99%以上同一の配列を除去。
dedupe.sh in=X.fa out=Y.fa minidentity=99
indelは許容されない。indelも考慮するならe flagを使用する。
・200bp以上同一の配列を1塩基のミスマッチまで許容して除去。
dedupe.sh in=X.fq pattern=cluster%.fq ac=f am=f s=1 mo=200 c pc csf=stats.txt outbest=best.fq fo c mcs=3 cc dot=graph.dot
duplication sequenceというよりアンプリコンシーケンスデータ向けのコマンド。似た配列はクラスタリングして出力され、同時に長さとクオリティがベストの配列は別ファイルに出力される。
・PacBioの16S全長シーケンスデータのクラスタリング。
1、全長読めてない配列を除去。
reformat.sh in=reads_of_insert.fastq out=filtered.fq minlen=1420 maxlen=1640 maq=20 qin=33
- minlength=0 (ml) Reads shorter than this after trimming will be discarded. Pairs will be discarded only if both are shorter.
- maxlength=0 If nonzero, reads longer than this after trimming will be discarded.
- minavgquality=0 (maq) Reads with average quality (after trimming) below this will be discarded.
- qin=auto ASCII offset for input quality. May be 33 (Sanger), 64 (Illumina), or auto.
1420-bp以下のリードと1640-bp以上のリード(アーティファクト)を除去する。また平均クオリティが20以下のリードも除去する。
2、99%以上同一の配列をクラスタリング。
dedupe.sh in=filtered.fq csf=stats_e26.txt outbest=best_e26.fq qin=33 usejni=t am=f ac=f fo c rnc=f mcs=3 k=27 mo=1420 ow cc pto nam=4 e=26 pattern=cluster_%.fq dot=graph.dot
- usejni=f (jni) Do alignments in C code, which is faster, if an edit distance is allowed. This will require compiling the C code; details are in /jni/README.txt.
- absorbmatch=t (am) Absorb exact matches of contigs.
- absorbcontainment=t (ac) Absorb full containments of contigs.
- findoverlap=f (fo) Find overlaps between contigs (containments and non-containments). Necessary for clustering.
- renameclusters=f (rnc) Rename contigs to indicate which cluster they are in.
- ow=f (overwrite) Overwrites files that already exist.
- cc=t (canonicizeclusters) Flip contigs so clusters have a single orientation.
- pto=f (preventtransitiveoverlaps) Do not look for new edges between nodes in the same cluster.
- minclustersize=1 (mcs) Do not output clusters smaller than this.
- cluster=f (c) Group overlapping contigs into clusters.
- numaffixmaps=1 (nam) Number of prefixes/suffixes to index per contig. Higher is more sensitive, if edits are allowed.
- maxedits=0 (e) Allow up to this many edits (subs or indels). Higher is slower.
4塩基オーバーラップの27merのseedを使い、99%以上同一で(e=26)1420塩基以上オーバーラップする配列をクラスタリングする。
コマンドが多いので、2回に分けます。
引用
BBMap (aligner for DNA/RNAseq) is now open-source and available for download.
BBMap (aligner for DNA/RNAseq) is now open-source and available for download. - SEQanswers
BBMerge - Accurate paired shotgun read merging via overlap.
Bushnell B, Rood J, Singer E
PLoS One. 2017 Oct 26;12(10):e0185056. doi: 10.1371/journal.pone.0185056. eCollection 2017.
BIostars ダウンサンプリング
https://www.biostars.org/p/168178/