Seaviewバージョン5(*1)
分子配列データからマルチプルアライメントと系統樹構築を行うマルチプラットフォーム・プログラム、Seaviewバージョン5を紹介する。Seaviewは、配列データベースへのネットワークアクセス、任意のアルゴリズムによるアラインメント、PhyMLを用いたparsimony(節約)とdistance(距離行列)、maximum likelihood(最尤)の系統樹の構築、ルート有りと無し、バイナリ、マルチ分岐の系統樹の表示、印刷、クリップボードへのコピー、SVGファイルへのコピーなどができる。Seaviewはグラフィカルユーザーインターフェースを提供するプログラムである。Seaviewバージョン5では、遺伝子ツリーをリファレンス種ツリーと照合し、この照合を用いて遺伝子ツリーのルートや再配置を行う機能が導入された。Seaview は http://doua.prabi.fr/software/seaview から自由に利用できる。
Seaviewバージョン4(*2)
分子配列データからのマルチプルアラインメントと系統樹再構築は、多くの分子進化解析にとって重要な作業である。これらの作業には、完全な手順の一部を実行する複数のプログラムを連続して使用する必要があり、これらのプログラム間で配列や樹を転送するために、しばしば面倒でエラーを起こしやすい一連のデータ再フォーマットが必要となる。コンピュータプログラムSeaViewとPhylo_winは、多重配列アライメントと系統樹再構築を行うためのグラフィカルユーザーインターフェースの先駆者である(Galtier et al. 1996)。これらのプログラムは広く使われてきたが、最近開発された最尤法の系統樹推定を利用できなかった。SeaViewバージョン4は、公開データベースからのネットワークベースの配列抽出から、最新のアラインメントと最尤法の系統樹構築アルゴリズムを用いたツリー構築と表示まで、相同DNA配列またはタンパク質配列の完全な系統解析を行うことができるプログラムである。
インストール
macosでテストした(mac mini2018使用)。apple silicon macでも動作はすることを確認。
HPからダウンロードする。
https://doua.prabi.fr/software/seaview

seaviewはGUIなしでコマンドラインから実行可能だが(TOPより)、ここではGUI版を使う。
実行方法
ダウンロード後そのまま開く。

Fileから配列を読み込む。

ここではOPENからmulti-fastaの配列を読み込んだ。

ここではSSU rRNA配列を使用。
基本的な機能を見てみる。
SeaViewは複数配列アラインメントエディターでもある。1つまたは複数の配列の1つ以上のギャップを同時に追加または削除したり、配列をさらに追加・編集出来る。
配列選択はマウスでクリックする(複数可)。再度クリックすると選択は解除される。選択した配列の数によって、利用可能なメニューは変化する。配列を編集するには、1つだけ配列を選択して、Edit sequenceを選択する。

別ウィンドウで編集後、Applyで適用される。配列名も変更できる。

配列を選択して、逆鎖、相補鎖、U <=> T変換も可能。

Select allで全配列を選択できる。Commnad + Aでも可能。全選択の解除は右クリックする。

Edit => Load sequence

配列名と配列を入力後、Add to alignmentをクリック。

新しい配列が追加された。

配列を1つ以上選択後、Delete sequenceで配列を削除。

ギャップ(-)だけの配列を自動で削除。

Edit => Dot plot

(3つ以上配列を選択しているとアクティブにならない)
別ウィンドウで表示される。拡大縮小、スクロール、PDF書き出しなどに対応している。

ドットプロット解析を任意の2つの配列間で行うことができ、アラインメントアルゴリズムが配列類似性の高い領域を見逃していないかを視覚的にチェックできる。
Edit => Consensus sequence

選択した配列間のコンセンサス配列が追加される。
コンセンサスのパラメータはPropsから行う。

PropsでAllow seq. editingにチェックをつけると、直接配列を編集できる。

Align
多重整列を実行する。プログラムやパラメータはoptionから選択できる。デフォルトはclustal omega。

Align allで全配列間を整列(多重整列)する。Align selected sequencesで選択した配列のみ。

SeaViewは多重配列アラインメントに外部のプログラムを使用している。2つのプログラムClustalW version 2とMuscleが利用できる。プログラムのデフォルトのパラメータ値は、ほとんどのケースでうまく動作するように選ばれている。
De-align selectionで全てのギャップを除去し、整列する前に戻せる。

Props => View as proteinでタンパク質に翻訳表示できる。

(選択を解除すると元に戻る)
Props => Font sizeでフォントサイズを変更できる。

Props => Statistics

特定の配列セットだけ選択したい時は、前もってSpecies => Create groupからグループを作っておく。

E.coliなどグループを示す名前をつけて保存する。

グループを作っておくと、選択解除後でも素早く再選択できる。

(Delete groupでグループ削除)
Create Foolerでコメント用の行を追加できる。

コメント行には自由に文字を書き込める。

(Deleteでコメント削除)
Trees
parsimony tree、distance tree、phyML(maximum likelihood) treeの系統樹の計算をお行える。前もってAlignから多重整列を実行しておく。

Parsimony
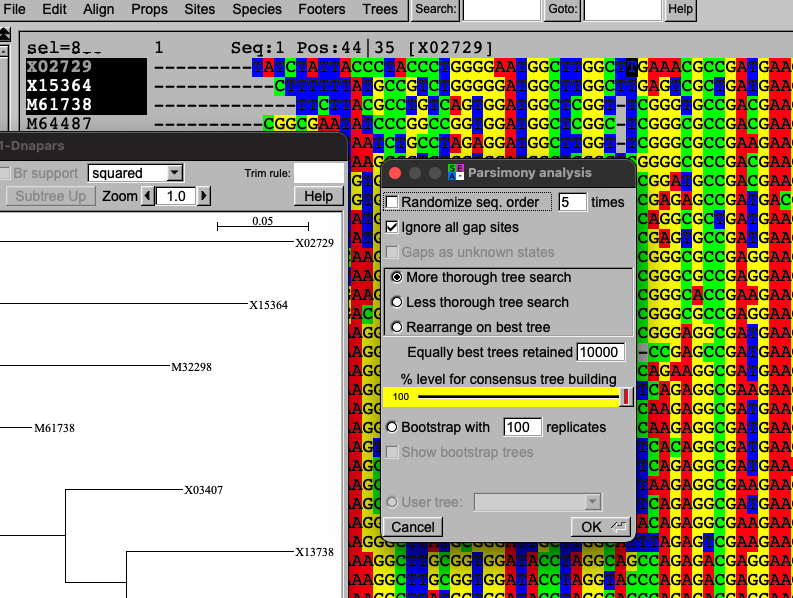
DnaparsとProtparsプログラムから抽出したコード(Phylip version 3.52, Felsenstein 1993)を使って、最大節約法の系統樹を計算できる。パーシモン計算は部位のブートストラップ再サンプリングと組み合わせることができ、配列の入力順序をランダムに変更した後、ユーザーが選んだ回数を繰り返すことができる。ユーザーが指定したツリーのパーシモンスコアも計算できる。(論文より)
Distance methods

2つの距離ベースの系統樹再構築法が含まれている: Neighbor-Joining (Saitou and Nei 1987; Studier and Keppler 1988)とBioNJ (Gascuel 1997)。これらは、様々なヌクレオチドおよびタンパク質配列のペアワイズ距離に適用でき、ブートストラップ再サンプリングと組み合わせてブランチサポート推定を行うことができる。ヌクレオチドレベルの距離は、観察ダイバージェンス(Observed)、Jukes Cantor (JC)、Kimuraの2パラメータ(K2P)、Hasegawa-Kishino-Yano、LogDet (Lake 1994)、タンパク質コード配列のLiの非同義(Ka)と同義(Ks)距離(Li 1993)である。ギャップを含む部位はデフォルトでペアワイズ距離計算から除外される。(論文より)
Distance treeの出力例

ツリーのプロットは、枝長、枝のサポート値(通常、ブートストラップスコアまたは近似尤度比検定確率)、バイナリまたはマルチ分岐、rootedまたはunrootedを表示できる。
PhyML

SeaViewは最尤系統樹の再構築には独立したプログラムとしてPhyML version 3 (Guindon and Gascuel 2003)を利用している。ツリー構築は、全配列または選択した配列、全配列部位または選択した配列部位に適用することができる。近似尤度比検定 (Anisimova and Gascuel 2006)、またはブートストラップ再標本 (Felsenstein 1985)によって枝支持を推定できる。(論文より)
Rerootするには、rerootを選択後、■のアイコンをクリックする。

Fullに戻すと、Br(branch length)とBr support(Bootstrap support)のチェックマークがアクティブになって数値をプロットできるようになる。
論文より
- SeaViewは特にMEGA 4に匹敵し、複数配列のアラインメントと系統樹再構築と表示のための精巧なグラフィカルユーザーインターフェースを提供している。
- SeaViewは、MEGAに比べて一対の距離計算の汎用性が低く、中立性や分子検定などの機能が欠けているが、すべての主要なコンピュータプラットフォームで利用でき、PhyMLによる最尤ツリーの再構成が可能であるという点でユニークである。
- SeaViewバージョン4は、すべてのユーザーが無償で利用でき、ユーザーインターフェースが系統解析に関わる概念的なステップをグラフィカルに表現しているため、分子系統学を教える上で特に価値がある。
- SeaViewはまた、ファイル形式やプログラムオプションに関する多くの技術的な詳細に直面することから解放されるため、系統樹再構築を時々利用するユーザーにとっても有用である。このようにSeaViewは塩基レベルとタンパク質レベルの両方で最尤解析を行い、最新の進化モデルを実装し、統計的枝支持を計算するので、熟練した系統学者にとっても有用であると期待される。
引用
1
Seaview Version 5: A Multiplatform Software for Multiple Sequence Alignment, Molecular Phylogenetic Analyses, and Tree Reconciliation
Manolo Gouy, Eric Tannier, Nicolas Comte, David P Parsons
Methods Mol Biol. 2021;2231:241-260
2
SeaView version 4: A multiplatform graphical user interface for sequence alignment and phylogenetic tree building
Manolo Gouy, Stéphane Guindon, Olivier Gascuel
Mol Biol Evol. 2010 Feb;27(2):221-4
3
SEAVIEW and PHYLO_WIN: two graphic tools for sequence alignment and molecular phylogeny
N Galtier, M Gouy, C Gautier
Comput Appl Biosci. 1996 Dec;12(6):543-8