微生物叢の解析は、健康や科学にとって重要な意味を持つ。これらの解析では、16S/18S rRNA遺伝子シーケンスを利用して分類群を同定し、種の多様性を予測する。しかし、微生物叢データを解析するための利用可能なツールのほとんどは、適切な実装のために熟練したプログラミングスキルと深い統計的知識を必要とする。ロングリードアンプリコンシーケンスは、より正確な分類群の予測につながり、急速に普及しつつあるが、実務者が簡単に利用できる解析ツールはない。ここでは、微生物相アンプリコンシーケンス解析のためのGUIツールであるMOCHIを発表する。MOCHIは、配列の前処理、分類の割り当て、異なる豊富な種の同定、種の多様性と機能の予測を行う。16S/18S rRNAの部分配列や16S rRNA全長配列の分類数表やFASTQを入力とし、リアルタイムで解析を行う。リアルタイムで解析を行い、表形式とグラフ形式の両方でデータを可視化する。MOCHIはローカルにインストールすることも、ウェブツールとして https://mochi.life.nctu.edu.tw からアクセスすることもできる。
ショートリードのアンプリコンだけでなく、ロングリードのアンプリコンにも対応しています。
https://mochi.life.nctu.edu.tw/にアクセスする。

1、Sequence Preprocessingタブ
Sequence PreprocessingのStep1を選択する。

fastq.gzかfq.gzのシークエンシングデータをアップロードする。ここではExample sequences ボタンからサンプルファイルをダウンロードし、そのファイルをアップロードした。
パラメータを設定する。シングルエンドまたはペアエンド、もしくはロングリードかどうか、プライマーの種類、計算スレッド数を指定する。0の場合、利用可能なすべてのコアが使用される。

STARTをクリックする。ジョブIDは解析結果を取得するために使用され、2週間サーバーに保存される。
Sequencing counts summary

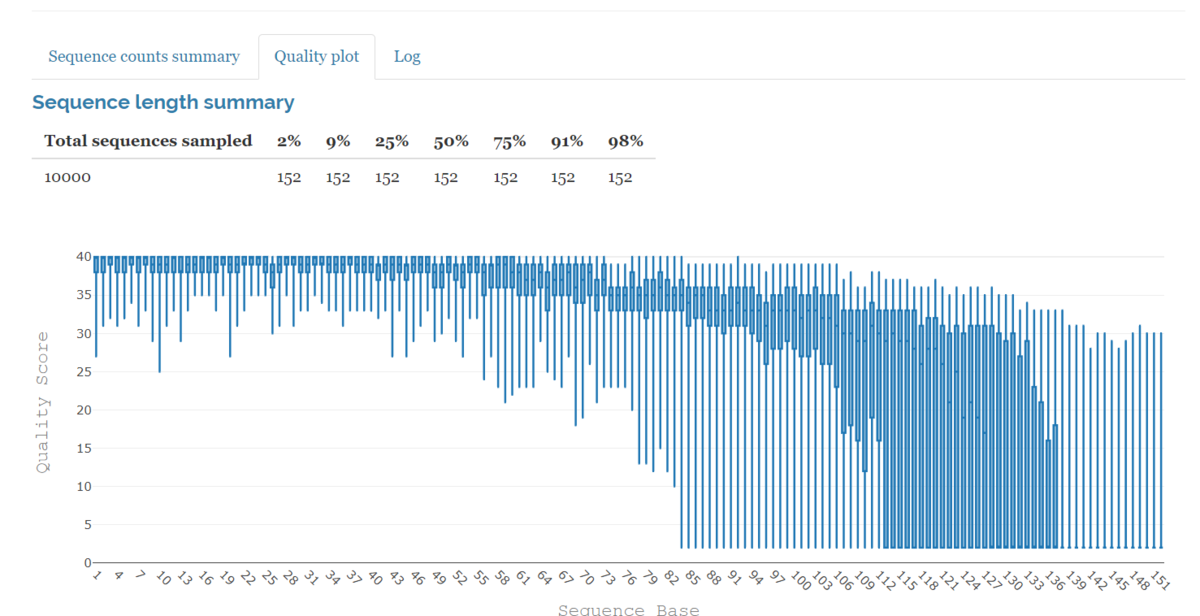
Quality plot
Step 2. Sequence denoisingを選択する。Step 1.で選択したシーケンスの種類によって、設定内容が異なる。

- トリミングの開始位置、終了位置、Quality Scoreを設定する。開始位置より下、終了位置より上の塩基対は切り落とされる。例えば、開始位置を5、終了位置を120に設定すると、5〜120bpの配列が得られる。また、終了位置より短いリードは破棄される。End positionが0の場合、切り捨てや長さのフィルタリングは行われない。
- Quality scoreについては、品質スコアが指定値以下のヌクレオチドが切り捨てられる。
- キメラリードとは、複数の親配列に由来する配列を意味する。キメラリードは一般にコンタミだが新規配列と解釈される可能性がある。が、実際はアーチファクトである。Chimeric reads filteringの値が高いほど、より多くのキメラリードが解析に使用される。ほとんどの場合、1がデフォルト値。
(マニュアルより)
パラメータを決めたらSTARTをクリックする。
出力例

Filter info

Sequence info
Rarefraction plot

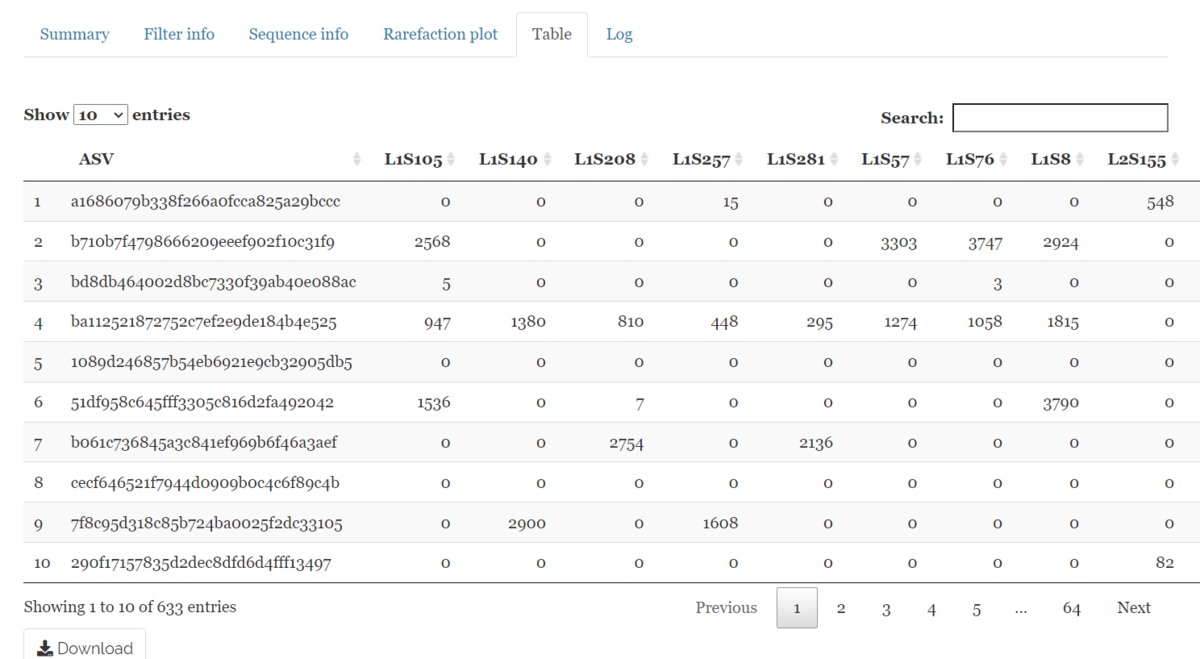
すべてのサンプルにおける各ASVのリードカウント。
Step 3. Taxonomy classificationを選択する。Step 1.で選択したシーケンスの種類によって、設定内容が異なる。

分類群を予測するためのデータベース(Silva、Greengene、PR2)を選択する。最新のデータベースがサーバーからダウンロードされ取り込まれる。

指定された値の範囲外のリファレンス配列は破棄される。デフォルト値は、ノイズ除去された配列の最小および最大の長さになる。長さフィルタリングを無効にするには値をゼロに設定する。

最小のデフォルト値はdenoised-sequencesの中央値 - 100、最大の デフォルト値はdenoised-sequencesの中央値 + 100。
出力例
ASVと割り当てられた分類群が表示される。

分類テーブル、ASVテーブルをダウンロードできる。
2、Taxonomic analysisタブ

メタデータ、分類テーブル、ASVテーブル(OTUテーブル)のファイルをアップロードする。分類テーブル、ASVテーブルはSequence Preprocessingタブで得られる(MOCHI/QIIME2形式指定)。18Sなら18Sにチェックを付ける。
パラメータを決めたらSTARTをクリックする。
出力例
Taxonomic table

K はあるレベルの分類群の数を示す。表の右側にはリードカウントが表示される。これらは選択されたグループによって分類されている。
Taxonomic barplot
全サンプルの分類群のパーセンテージを示すインタラクティブなバープロット。

下のメニューのメタデータによって結果をグループ化できる。このメタデータはユーザーが提供したテキストに依存している。body.siteを選択。
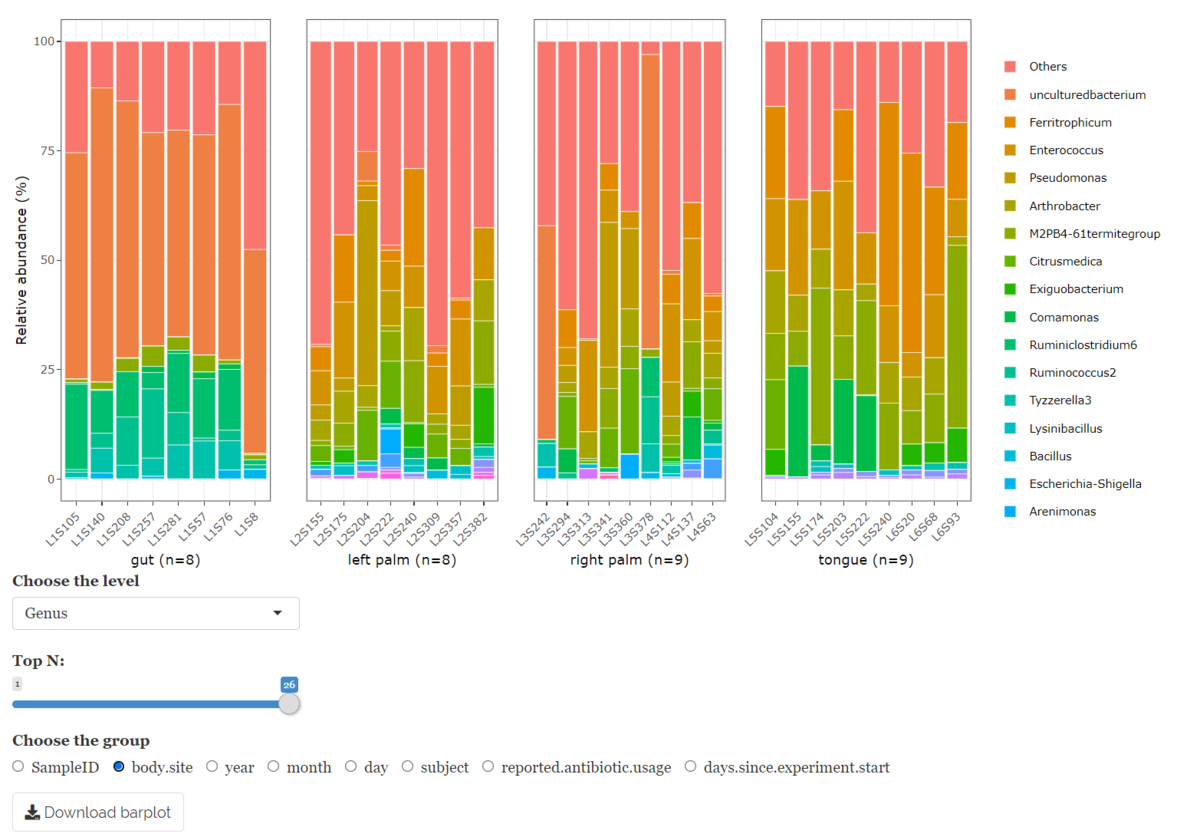
N の値を指定すると、プロットは各サンプルで比較的多い上位 N 種の分類群の和が表示される。例えばN = 2 の場合、サンプル A とサンプル B の上位 2 種類の豊富な分類群が「taxa_1 and taxa_2」、「taxa_1 と taxa_3」であった場合、プロットではtaxa_1、taxa_2、taxa_3 の相対的な存在量が示される。(マニュアルより)
Taxonomic heatmap
log10変換されたパーセンテージを示すインタラクティブなヒートマップ。ゼロの対数を取らないようにするため、変換前にすべてのパーセント値に小さな値0.01が足されている。

genusレベル、subjectでグループ化。

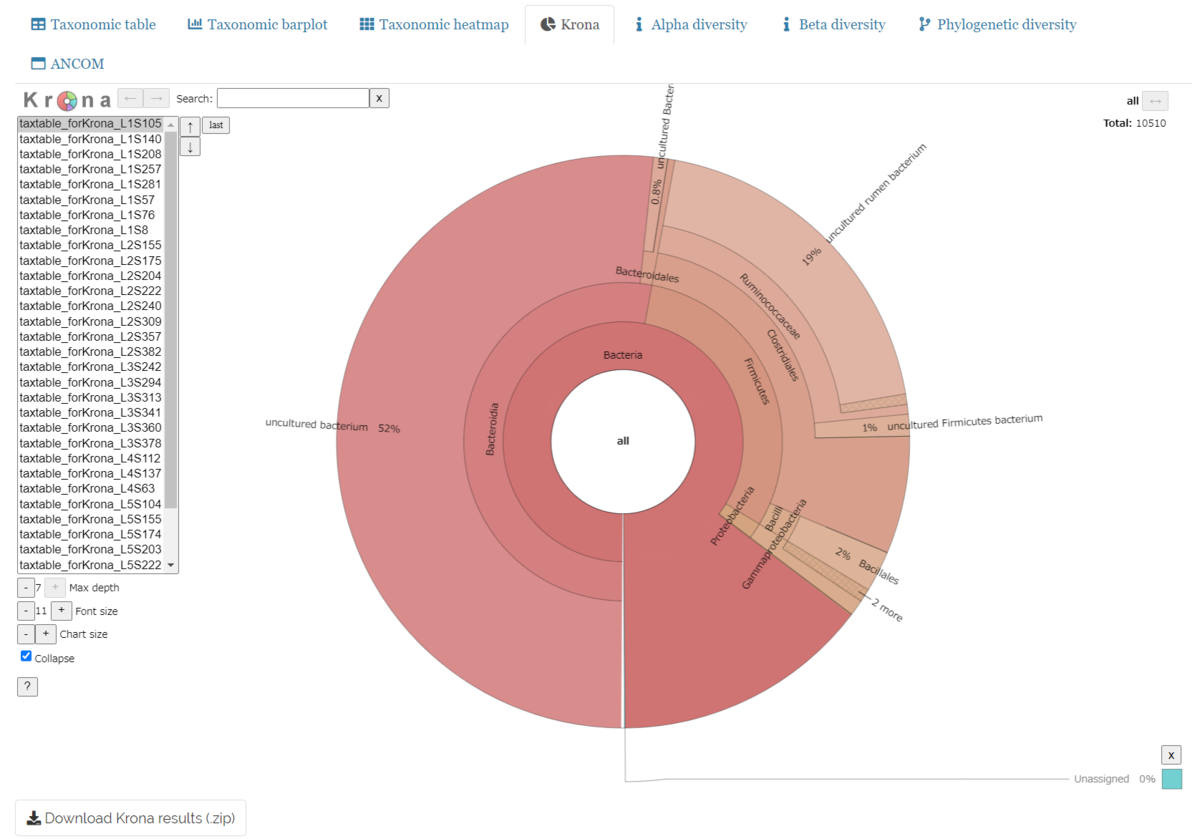
Krona
Alpha diversity
6つのアルファ多様性インデックスの値;ACE, Shannon diversity, InvSimpson diversity, Shannon evenness, and Simpson evennessで示される。


Shannon diversityを選択、Yearでグループ化。ANOVA(パラメトリック手法)または Kruskal-Wallis(ノンパラメトリック手法)を選択し、指数の分布が有意であるかどうかを検定する。

Beta diversity
サンプル間の種の多様性を評価するための指標。MOCHIではBray-Curtis指数を用いている。

ヒートマップ上にカーソルを置くと、種間の距離が表示される。

ヒートマップに表示される数値は、元の数値の自然対数を0.01倍したもの。
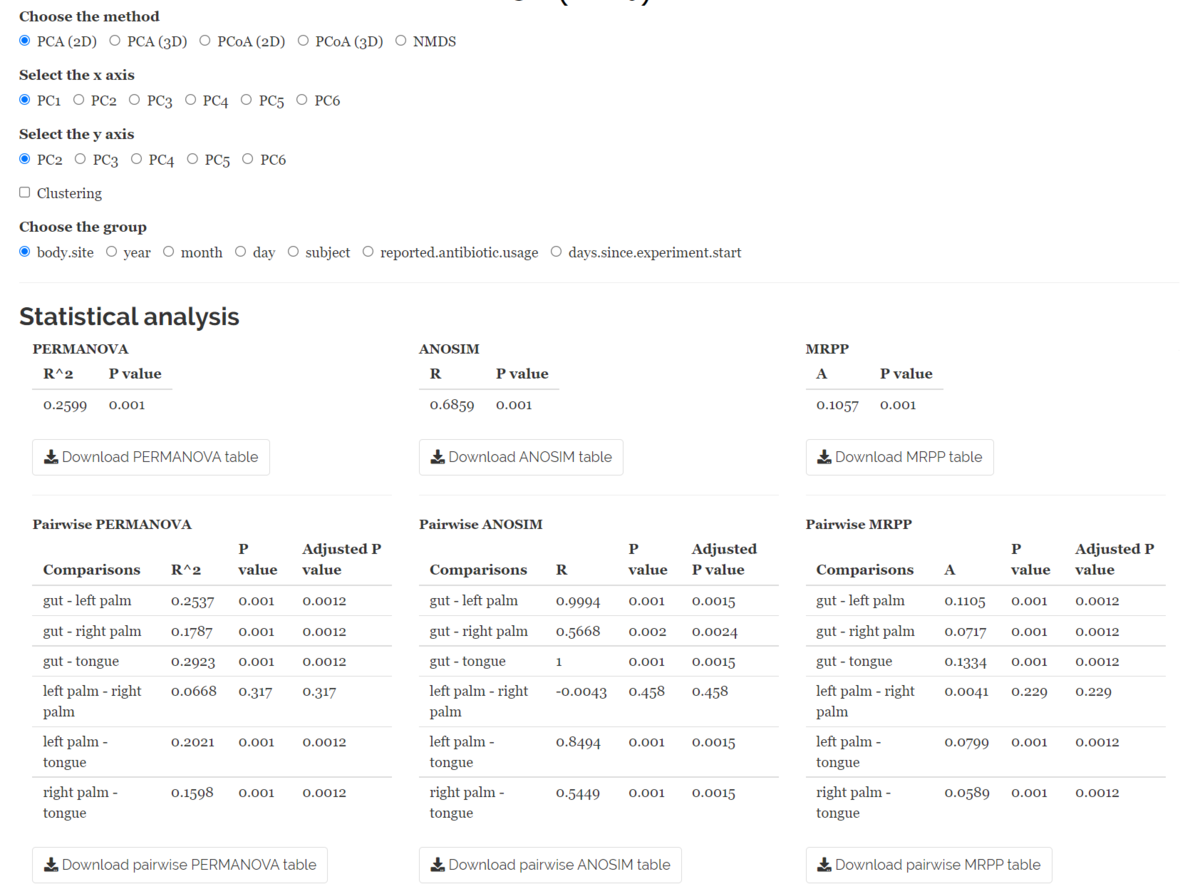
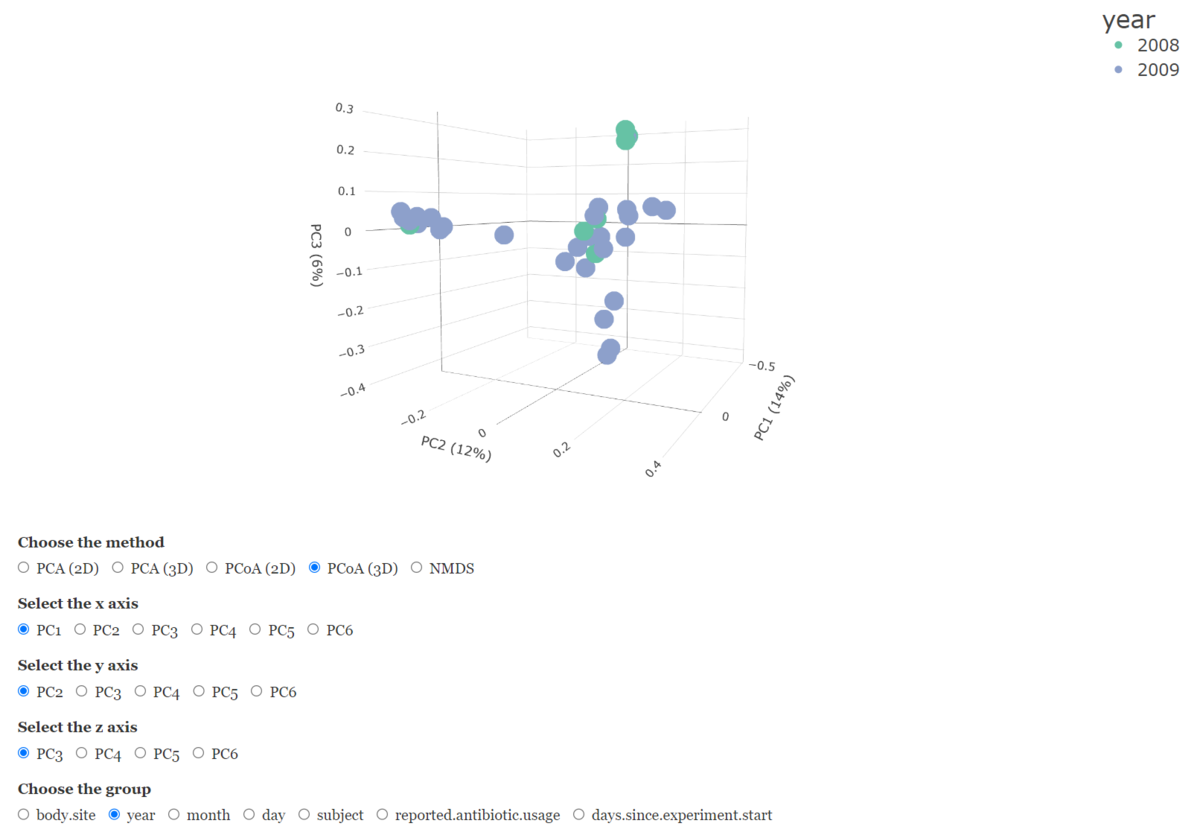
ベータ版の多様性を可視化するために、PCA (Principal Component Analysis, 2D & 3D), PCoA (Principal Co-ordinates Analysis,2D & 3D) 、NMDS (Non-metric Multidimensional Scaling)の3種類の次元削減法が提供されている。

PCoA 3D、year、PC1, PC2, PC3。
Statistical analysis
β多様性がグループ間、あるいはペア間で有意に異なるかどうか調べるために、PerMANOVA(順列多変量解析)、ANOSIM(類似性解析)、MRPP(多重応答並べ替え法)の3つの統計手法を提供している。

Phylogenic diversity
種間の遺伝的な差異を定量的に把握するための多様性の指標。シーケンスファイル(.qza)をアップロードする。配列の前処理を完了している場合は、「配列の前処理-分類」の後にファイルをダウンロードする。

詳細はマニュアル参照。
3、Function analysis
微生物相の機能を予測するためのデータベースFAPROTAXを利用した機能解析。

メタデータ、分類テーブルのファイルをアップロードする。分類テーブルはSequence Preprocessingタブで得られる(MOCHI/QIIME2形式指定)。
出力例
機能アノテーションテーブル。各サンプルの機能タイプを表示する。

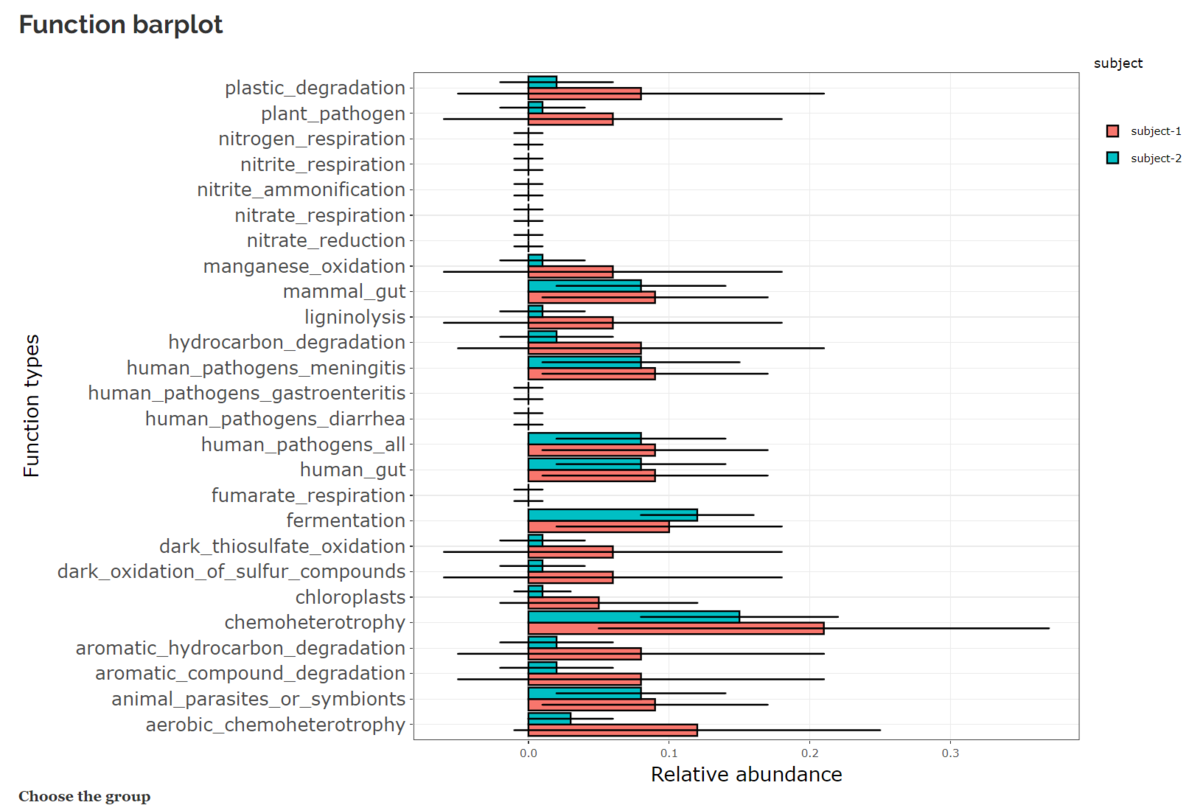
Function plot
各機能のリード数がメタデータに基づいてグループ化されている。

subjectに変更。
引用
MOCHI, a comprehensive cross-platform tool for amplicon-based microbiota analysis
Jun-Jie Zheng, Po-Wen Wang, Tzu-Wen Huang, Yao-Jong Yang, Hua-Sheng Chiu, Pavel Sumazin, Ting-Wen Chen
Bioinformatics. 2022 Jul 25;btac494