多様な生物種のゲノム配列がますます豊富になる時代において、タンパク質をコードする遺伝子レパートリーの質を評価することは極めて重要である。最先端のゲノムアノテーション評価ツールは、遺伝子レパートリーの完全性を測定するが、遺伝子の過剰予測やコンタミネーションなど、他のタイプのエラーは検出できない。
OMArkは、クエリプロテオームと生命のツリー全体にわたって事前に計算された遺伝子ファミリーとの間の、アライメント不要の高速配列比較に依存するソフトウェアである。OMArkは完全性だけでなく、近縁種と比較した遺伝子レパートリー全体の一貫性も評価する。また、汚染の可能性が高い事象も報告する。
OMArkをシミュレーションデータで検証した後、1805のUniProt Eukaryotic Reference Proteomesの解析を行い、品質尺度に基づくプロテオームの比較と優先順位付けの有用性を示した。特に、59のプロテオームでコンタミネーションの強い証拠を発見し、断片化されたゼブラフィンチのプロテオームをリファレンスとして使用した結果、鳥類の遺伝子アノテーションでエラーが伝播していることを特定した。
OMArkはGitHub (https://github.com/DessimozLab/OMArk)、PyPi上のPythonパッケージ、https://omark.omabrowser.org/ のインタラクティブオンラインツールとして利用できる。
https://lab.dessimoz.org/blog/2022/12/12/omark-to-evaluate-proteome-quality より
多くの研究は、ゲノムアセンブリーから予測されるタンパク質コード遺伝子レパートリーの「プロテオーム」に直接依存して比較を行っている。そうすることで、すべてのゲノムの予測された遺伝子コンテンツは均質であり、現実を正確に反映しているという仮定に依存している。しかし実際には、この仮定が満たされることはほとんどなく、報告されたプロテオームにおいてタンパク質コード遺伝子が欠落していたり断片化されていたり、遺伝子予測ツールによって非コード配列が誤ってコード遺伝子としてアノテーションされていたり、報告された配列の中に他の生物種からの汚染が誤って含まれていたりすることがしばしばある。
開発した新しい手法OMArkは、プロテオームの質の様々な側面を簡単かつ包括的に測定する方法を提供する。すなわち、遺伝子レパートリーの完全性、含まれる遺伝子の分類学的レベルでの一貫性、疑わしい遺伝子構造の有無、ドメイン間あるいはドメイン内のコンタミネーションの有無などである。さらに、既存の方法とは異なり、OMArkは参照データセットの手動選択に頼らず、代わりにテストプロテオームの最も可能性の高い分類学的分類(taxonomic classification)を自動的に特定する。このため、普遍的な参照データベースを用いて、生命のツリーにわたるあらゆるテストプロテオームを処理することができる。
1805の真核生物UniProtリファレンスプロテオームの大規模解析をOMArkで行い、不完全性、コンタミネーション、翻訳された非コード配列の混入のいずれかに起因する品質問題の明確なケースを検出することができた。最も極端なケースでは、8つの異なる種(菌類とバクテリア)からのコンタミネーションがある植物プロテオームを発見した。
2024/02/24
https://twitter.com/Why_NeverS/status/1760299559538901084
ここではまずOMArkのwebサーバーを紹介します。続いてローカルマシンで使用する手順も簡単に紹介します。
https://omark.omabrowser.org/にアクセスする。

目的のゲノムの各タンパク質コード遺伝子のFASTAファイルをアップロードする。圧縮ファイルは認識しない。さらに任意で対象種のNCBI分類IDを指定する。OMArk はこれを系統選択に使用する。さらに、FASTA ファイルにアイソフォームが含まれている場合、各遺伝子の全てのアイソフォームをリストしたファイルをアップする(任意)。

サブミットする。計算が終わるまでしばらく待つ必要がある。プレスリリースでは約30分で結果が得られるとある。
出力
結果は視覚化される。

論文の図1で、OMArkの出力について簡単に説明されている。以下、図1より。
”OMArkは主に2つの品質評価カテゴリーを提供する: 1) 完全性評価(種の系統の保存された祖先遺伝子セットとクエリープロテオームの重なりに基づく)。完全性評価では、クエリプロテオーム中の遺伝子をSingle copy、Multiple copy (Duplicated)、Missingに分類する。これはBUSCOのような手法に似ているが、複数コピーある遺伝子も考慮する。次に、2) 一貫性評価(Consistency assessmentの訳)、正しい系統の遺伝子ファミリーにクエリタンパク質が含まれる割合(Consistent)、正しくない系統の遺伝子ファミリーにクエリタンパク質が含まれる割合(ランダムに含まれるか(Inconsistent)、特定の生物種に含まれるか(Contamination))、そして遺伝子ファミリーに全く含まれないクエリタンパク質(Unknown)。
近縁種(UniProtリファレンスプロテオーム)の事前計算結果と自分のプロテオームの品質を比較することができる。右上のchange to comparison ~をクリックする。自分のプロテオームは赤色の"yeast"。
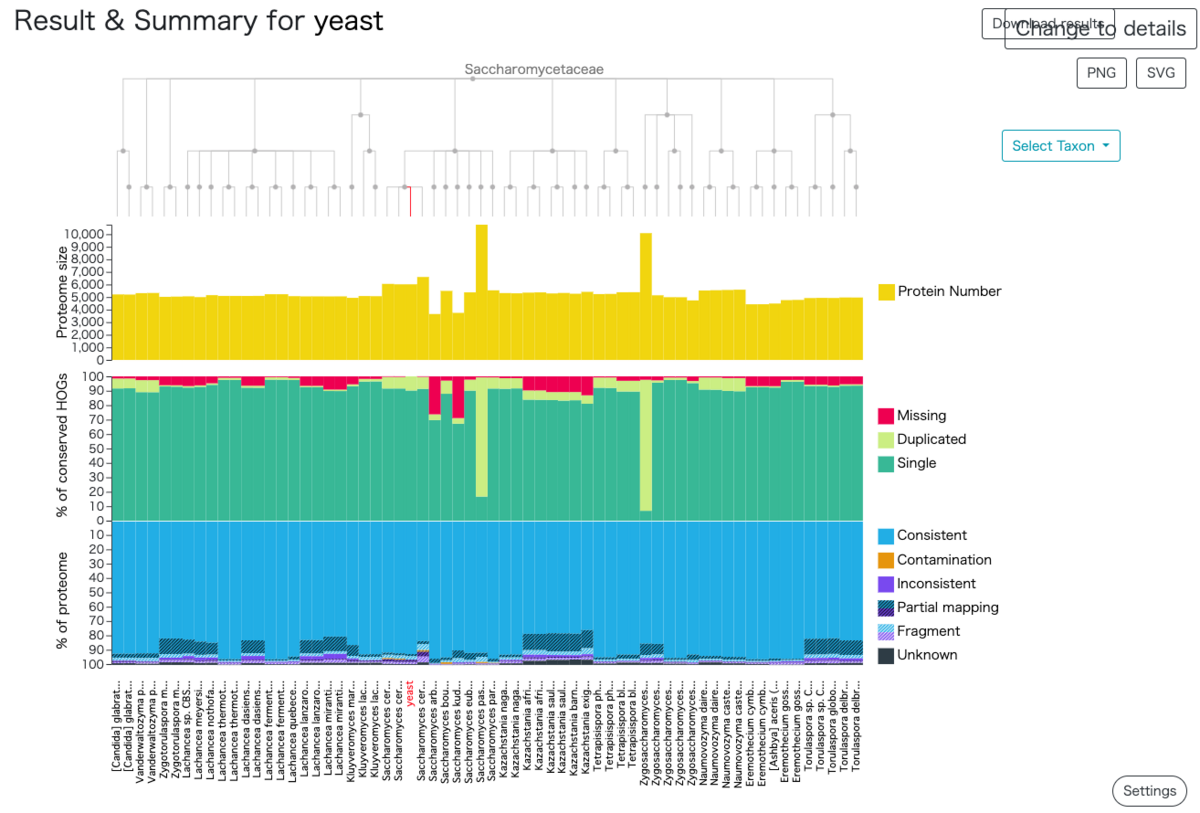
select taxonから他の分類群に切り替えて比較することもできる。グラフの要素をクリックするとそのゲノムの評価ページにジャンプする。
続いてローカルマシンへの導入と使用方法を確認する。
インストール
#PyPI(link)
pip install omark
#conda
git clone https://github.com/DessimozLab/OMArk.git
mamba env create --file OMArk/omark_env.yml
* 依存のPySAISが正常に導入できなかった。
データベース
全データベースと各グループを個別にダウンロードすることができる。
https://omabrowser.org/oma/current/
テストラン
OMArkは、OMAオルソロジーデータベースと、全タンパク質を遺伝子ファミリーに高速に配置するためのOMAmerソフトウェアに依存している。まずOMAmerをクエリタンパク質ファイルを指定して実行する。
git clone https://github.com/DessimozLab/OMArk.git
cd OMArk/example_data/
omamer search --db LUCA.h5 --query UP000005640_9606.fasta --out UP000005640_9606.omamer
空の出力フォルダを作成後、OMArkを実行する。
mkdir omark_output
omark -f UP000005640_9606.omamer -d LUCA.h5 -o omark_output
その他(レポジトリより)
- OMArkを正しく動作させるために、データベースは幅広い種をカバーしていることが推奨される。できるだけOMAデータベース全体から構築されたもの(LUCA.h5 と呼ばれる)の使用を推奨する。
- より限定された分類学的範囲(Metazoa, Viridiplantae, Primates)のデータベースを使用すると、OMArkによる汚染の検出や、この範囲外に属する種の配列の同定が制限される。
引用
Multifaceted quality assessment of gene repertoire annotation with OMArk
Yannis Nevers, Victor Rossier, Clément Marie Train, Adrian Altenhoff, Christophe Dessimoz, Natasha Glover
bioRxiv, Posted November 28, 2022
追記
Quality assessment of gene repertoire annotations with OMArk
Yannis Nevers, Alex Warwick Vesztrocy, Victor Rossier, Clément-Marie Train, Adrian Altenhoff, Christophe Dessimoz & Natasha M. Glover
Nature Biotechnology, Published: 21 February 2024
関連
参考
What are reference proteomes?