次世代シーケンサー(NGS)は遺伝子研究に革命をもたらしたが、生のシーケンサーリードの前処理は依然として重要なステップである。品質管理(QC)とトリミングのための既存のツールには限界があり、ワークフローが分断されている。本稿では、使いやすいインターフェースでQC解析、トリミング、フォーマット変換を統合した、包括的でユーザー中心のソフトウェアソリューションである123FASTQを紹介する。123FASTQは、既存のツールの利点を組み合わせると同時に、その限界にも対処している。洞察に満ちたビジュアライゼーションによるQC分析の改善、QC結果に基づく半自動トリミング推奨、さまざまなトリミングオプションを提供する。また、さまざまなファイル形式をサポートし、効率的なパフォーマンスを提供する。123FASTQは、NGSデータの前処理を簡素化することで、研究者がダウンストリーム解析に高品質のリードを利用できるようにする。クロスプラットフォームで、https://sourceforge.net/projects/project-123ngs/から利用できる。
manual
https://dl.adbioinformatics.net/NGSNeeds/myTools/123Fastq_v1.3_Manual.pdf
インストール
ubuntu20でテストした。
依存
- 123Fastq is written in java and capable to launch in Linux, Windows or macOS by using only one .jar file
https://sourceforge.net/projects/project-123ngs/
ダウンロードして解凍する。
マニュアルPDFも含まれている。
実行方法
コンソールでjarファイルを実行する。
java jar 123Fastq.jar
立ち上がった
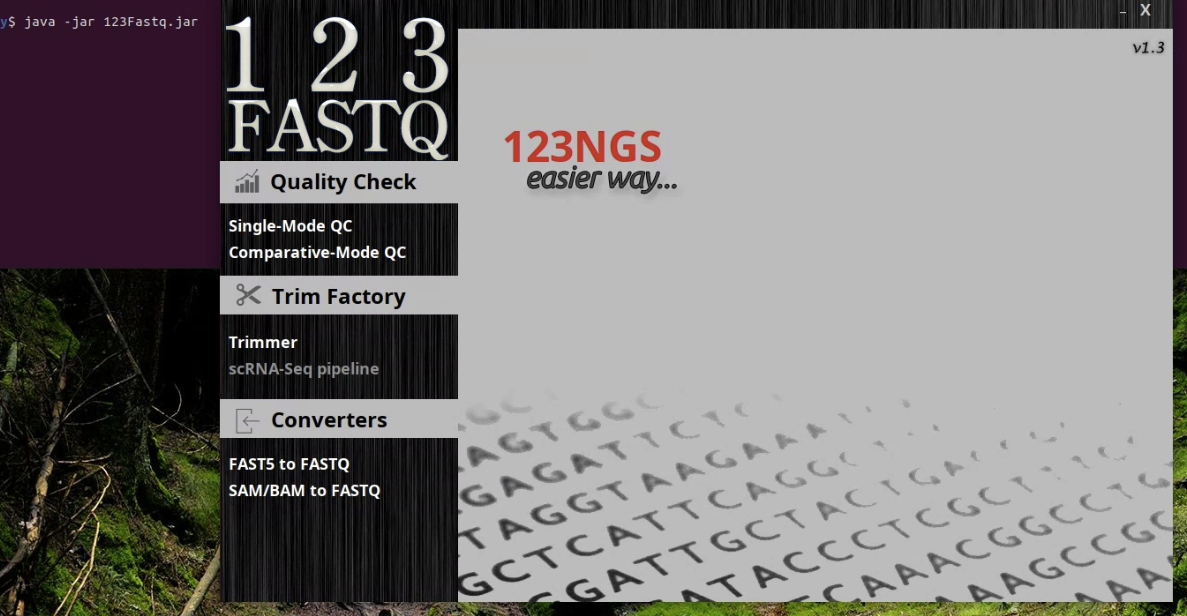
(ubuntu20.04 LTS Desktop版)
レポート機能はSingle-Mode QCとComparative-Mode QCに分かれている。前者はインポートしたファイルまたは一連のファイルに対する品質レポートを作成し、後者は2つのファイルまたは一連のファイルに対する品質レポートを作成する(ペアエンドデータやクリーニング前後のfastqなど)。
Single-Mode QC
左のメニューのSingle mode QCを選択するとfastqファイルを選択するパネルが表示される。fastqを選択すると、全リードがスキャンされる(複数ファイルも選択可能)。

スキャン後、レポートが作成・出力される。

FastQCと同様、表示項目を選択できる。

Adapter contentなどの項目もある。このツールは基本的にイルミナ用だがここで使用したのはMGIseqのfastqなので、illuminaデフォルトのアダプターの残存は一切検出されていない。

上の方にあるsave reportボタンをクリックすると、html形式でレポートを保存できる。
Comparative-Mode QC
左のメニューのComparative-Mode QCを選択するとfastqファイルを選択するパネルが二回繰り返し表示される。読み込むとスキャンが開始される。
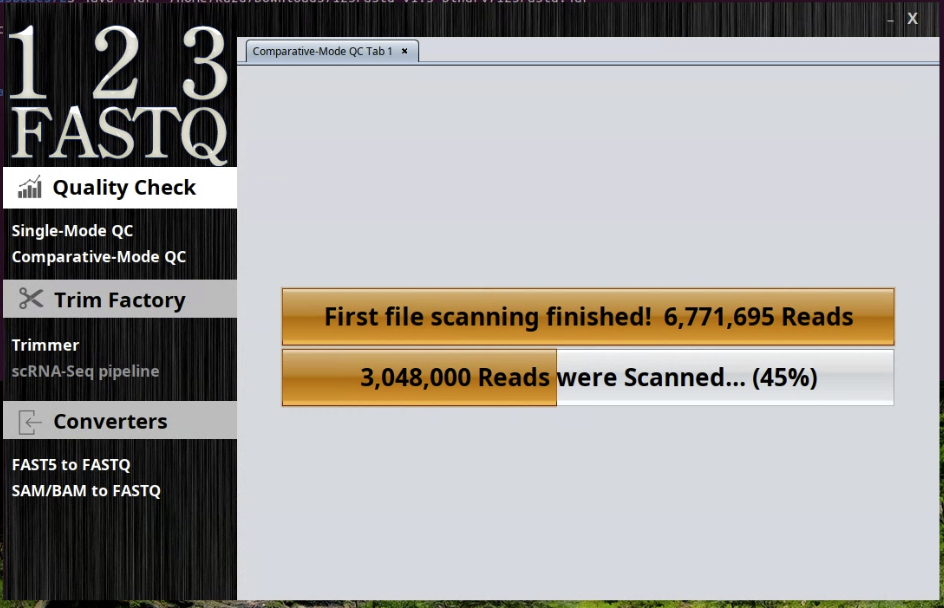
レポートはsingleと変わらないが、上のほうにfirstとsecondというボタンが表示されている。このボタンから2つのfastqのレポートを切り替えることができる。

fastqのレポート結果から、残存しているアダプターのトリミングに進めるようになっている。右上の(Paired-end) Trimをクリックする。
Trimmer
イルミナのアダプタートリミング機能。レポート結果からジャンプするか、左のメニューから選択する。

まずfastqを指定する。続いてエンコーディングや分析オプションを選択する。

スクロールする。

さらにスクロール

かなり細かい調整ができる。自動認識では除去できない自作アダプターを指定するボタンもあるが、2024/03現在のバージョンでは選択できない。
一番下で出力ディレクトリを指定して実行する。使用スレッドも選べる。
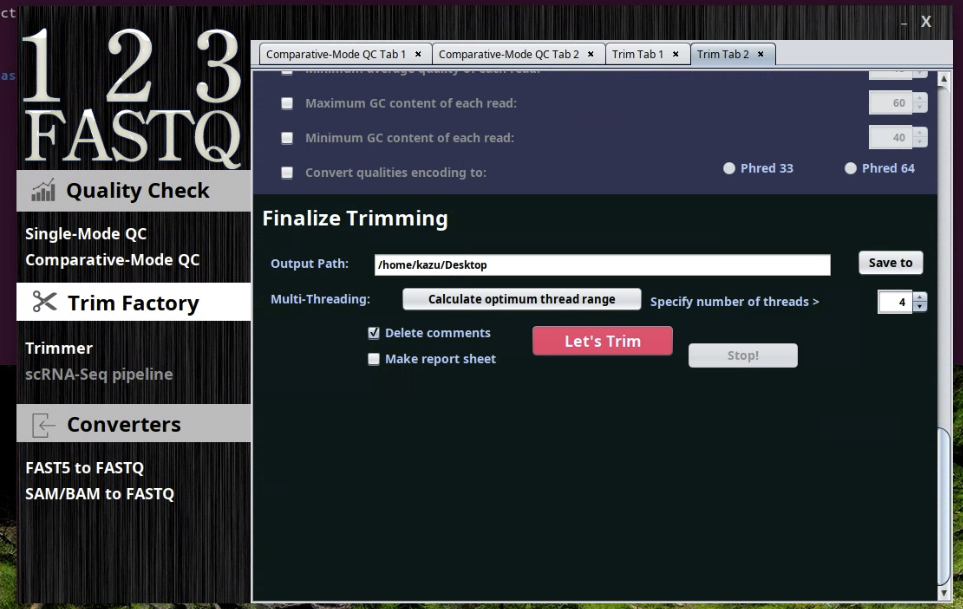
reportをオンしておくと、トリミング後に自動でQCがランされる。
Trimmer出力例
ペアエンドfastqをトリミングした場合、ペアが同期されたfastqと片方が脱落してシングルになったfastqがそれぞれ保存される。

他の機能
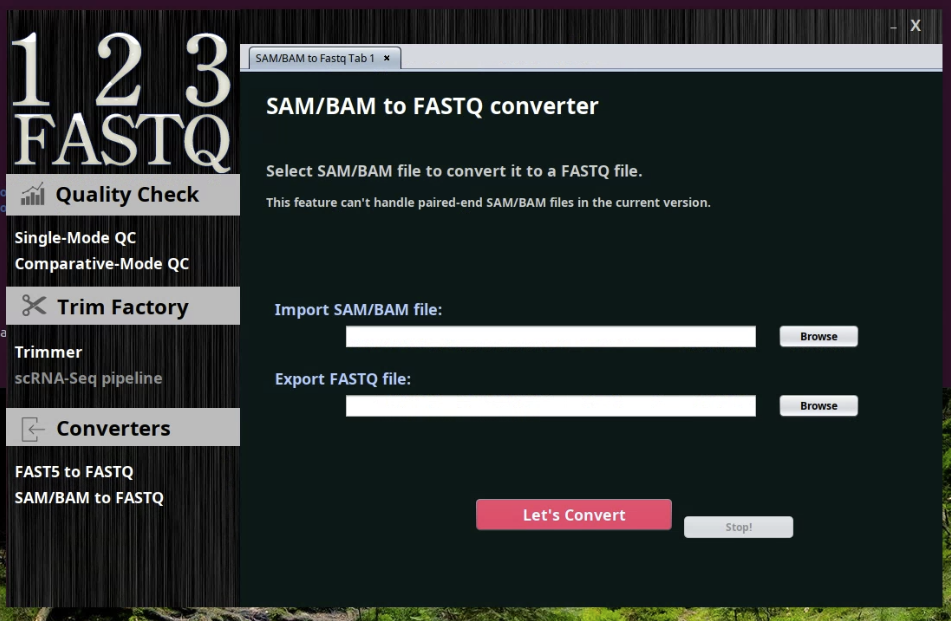
SAM/BAM => fastq変換
Fast5 => Fastq変換
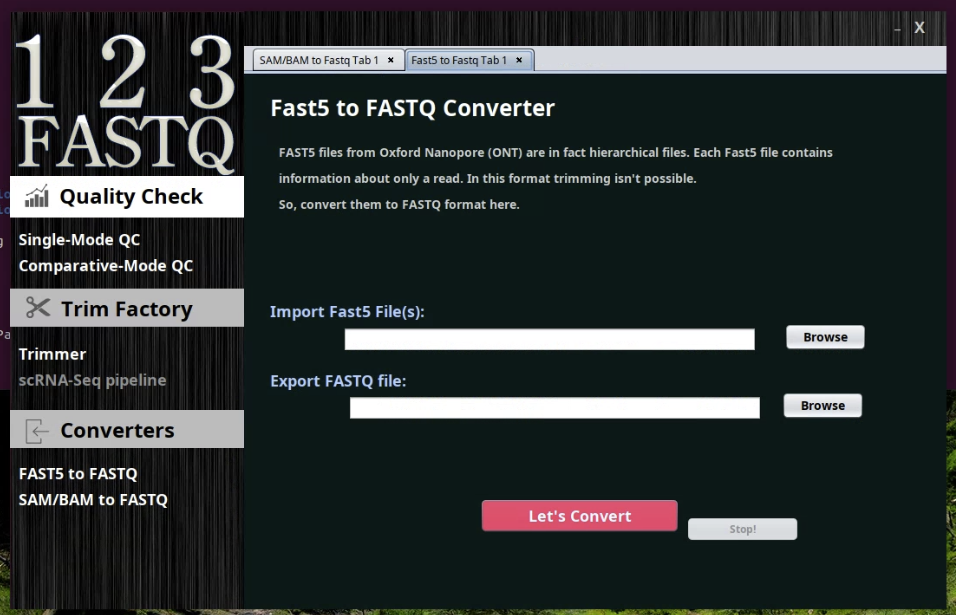
Oxford nanopore technologyシーケンサーの出力のFast5ファイルは、そのままではトリミングできない。そのため、 Fast5 ファイルをインポートしてFASTQ ファイルに変換する機能が用意されている。
その他
- Trim FactoryにはscRNA seq pipelineもあるが、ボタンが非アクティブになっている。論文のステータスがプレプリントの現在、まだ未実装なのだと思われる(マニュアルにも説明がない)。
引用
123FASTQ: an intuitive and efficient tool for preprocessing Illumina FASTQ reads
Milad Eidi, Samaneh Abdolalizadeh, Mohammad Hossein Nasirpour, Javad Zahiri, Masoud Garshasbi
bioRxiv, Posted March 10, 2024.