オックスフォード・ナノポア・テクノロジーズ(ONT)は、携帯可能で低コストなシーケンスの可能性から、生態学研究において人気を集めている第3世代のシーケンサー技術である。この技術はロングリードのシーケンスを得意とするが、アンプリコンのシーケンスにも応用できる。ONTの欠点は、生のリードの品質が低いことである。したがって、高品質のコンセンサス配列を生成することは、依然として課題である。本著者らは、ONTでシーケンスされたアンプリコンを、配列と長さの類似性に基づいて参照なしで選別し、確かなコンセンサス配列を構築するためのツール、Amplicon_sorterを紹介する。
GIthubより
- このスクリプトは、inputfile を読み込みでファイル内のすべてのリードのリード長ヒストグラムを作成する。
- その中から、minlength, maxlenght, maxreads に基づいて、リードの処理を開始する。処理結果は、同じ遺伝子のリードを含むgroup ファイルに保存される (例: group_1 には 18S リード、group_2 には COI リード、group_3 には ITS リードが含まれる)。
- group ファイルを処理して、種や属のレベルで遺伝子を選別し、別のファイルに保存する。(例: file_1_1.fasta は種 1 の 18S, file1_2.fasta は種 2 の 18S, ...)
- 各出力ファイルの末尾には、そのファイル中の配列のコンセンサスファイルが含まれる。
- グループごとに、そのグループのすべてのコンセンサス配列を含むファイルが作成される。全グループの全コンセンサス配列のファイルが作成される。
- 各出力ファイルの末尾には、グループごとに、そのグループのすべてのコンセンサス配列を含むファイルが作成される。
PDF manual
amplicon_sorter/Amplicon_sorter_manual.pdf at master · avierstr/amplicon_sorter · GitHub
インストール
mamba create -n amplicon_sorter python=3.9 -y
conda activate amplicon_sorter
python3 -m pip install python-Levenshtein
python3 -m pip install edlib
python3 -m pip install biopython
python3 -m pip install matplotlib
#本体
git clone https://github.com/avierstr/amplicon_sorter.git
cdamplicon_sorter/
> python amplicon_sorter.py -h
usage: amplicon_sorter.py [-h] -i INPUT [-min MINLENGTH] [-max MAXLENGTH] [-maxr MAXREADS] [-np NPROCESSES] [-sg SIMILAR_GENES] [-ssg SIMILAR_SPECIES_GROUPS] [-ss SIMILAR_SPECIES] [-sc SIMILAR_CONSENSUS]
[-sfq] [-ra] [-a] [-o OUTPUTFOLDER] [-ho] [-so]
AmpliconSorter: Sort amplicons based on identity and saves them in different files including the consensus.
optional arguments:
-h, --help show this help message and exit
-i INPUT, --input INPUT
Input file in fastq or fasta format
-min MINLENGTH, --minlength MINLENGTH
Minimum readlenght to process. Default=300
-max MAXLENGTH, --maxlength MAXLENGTH
Maximum readlenght to process. Default=No limit
-maxr MAXREADS, --maxreads MAXREADS
Maximum number of reads to process. Default=10000
-np NPROCESSES, --nprocesses NPROCESSES
Number of processors to use. Default=1
-sg SIMILAR_GENES, --similar_genes SIMILAR_GENES
Similarity to sort genes in groups (value between 50 and 100). Default=80.0
-ssg SIMILAR_SPECIES_GROUPS, --similar_species_groups SIMILAR_SPECIES_GROUPS
Similarity to CREATE species groups (value between 50 and 100). Default=estimate value from data
-ss SIMILAR_SPECIES, --similar_species SIMILAR_SPECIES
Similarity to ADD sequences to a species group (value between 50 and 100). Default=85.0
-sc SIMILAR_CONSENSUS, --similar_consensus SIMILAR_CONSENSUS
Similarity to COMBINE groups based on the consensus sequence (value between 50 and 100). Default=96.0
-sfq, --save_fastq Save the results also in fastq files (fastq files will not contain the consensus sequence)
-ra, --random Takes random reads from the inputfile.
-a, --all Compare all selected reads with each other. Only advised for a small number of reads (< 10000)
-o OUTPUTFOLDER, --outputfolder OUTPUTFOLDER
Save the results in the specified outputfolder. Default = current working directory
-ho, --histogram_only
Only creates a read length histogram.
-so, --species_only Only create species groups and sort to species level.
実行方法
NanoFiltなどの品質フィルタリングソフトで、入力ファイルから Q12 以上のリードをフィルタリングしてから使うことが推奨されている。
1、入力ファイルのリード長ヒストグラムを生成。期待されるサイズを指定する。
python3 amplicon_sorter.py -i infile.fastq –o outputfolder -min 650 -max 750 -ho
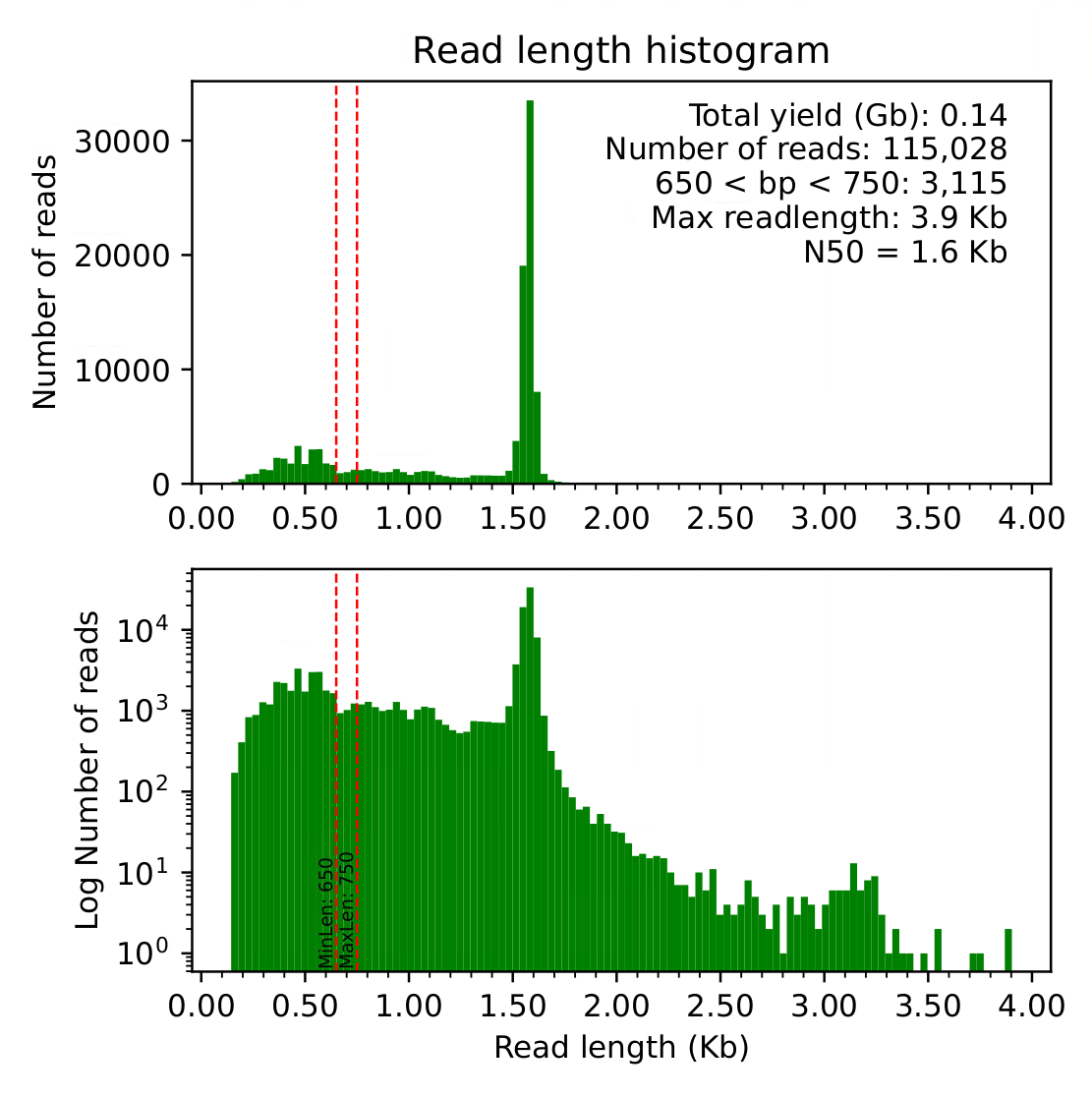
ユーザーが指定した期待されるサイズに赤の波線が付く。
2、750 bpの長さの1種のアンプリコンデータでサンプリングする。
入力ファイルをデフォルト設定で処理し、outputfolderフォルダに保存、8コアで実行、リードの最小長=700、リードの最大長=800、1000リードを使用。入力ファイルの700から800bpの間の最初の1000リードをサンプリングする。コマンドラインに-ra (random)オプションをつけると、700〜800bpのランダムリードを1000リードサンプリングする。
python3 amplicon_sorter.py -i infile.fastq -np 8 -min 700 -max 800 -maxr 1000
- -i Input file in fastq or fasta format
- -min Minimum readlenght to process. Default=300
- -max Maximum readlenght to process. Default=No limit
- -np Number of processors to use. Default=1
- -maxr Maximum number of reads to process. Default=10000
- -o Save the results in the specified outputfolder. Default = current working
3、700bpのアンプリコンと1200bpのアンプリコンの2種。2000リードサンプリングする。
python3 amplicon_sorter.py -i infile.fastq -np 8 -min 650 -max 1250 -maxr 2000 -o outdir
4、600~3000bpのアンプリコンを複数持つメタアンプリコンサンプル。種の数は不明、入力ファイルを30000リードサンプリングする。
python3 amplicon_sorter.py -i infile.fastq -o outputfolder -np 8 -min 550 -max 3050 -maxr 30000 -o outdir
最大リード数の20倍をランダムサンプリングすることで、低存在量の種を見つけることができる。
出力例

Githubより
- 95-96%以上類似した種を扱う場合は、Amplicon_sorterの設定を変更または微調整することが重要。
- サンプル中に94%以上類似した種がある場合、スクリプトはそれらを分離することが困難です。類似度の高い種は、グループ化され、2つ以上の近縁種の平均として低いコンセンサス配列が与えられることが多い。
引用
Amplicon_sorter: A tool for reference-free amplicon sorting based on sequence similarity and for building consensus sequences
Andy R Vierstraete, Bart P Braeckman
Ecol Evol. 2022 Mar 1;12(3):e8603
関連