2019 6/13 追記
2022 1/7 補足追加
エンリッチメント解析は、大量の遺伝子リストに関連する機能を決定し、生物学的過程を解釈する可能性を高めるための効率的かつ迅速な方法である(ref.1)。Biological processes(BP)、molecular functions(MF)、cell components(CC)に関する情報は、遺伝子オントロジー(GO)として知られる構造化された語彙に編成されている。新興のエンリッチメントツールの大部分(ref.2〜7)は、アノテーションリソースとしてGOを使用している。これらのリソースは一般的にヒトまたはモデル種を扱えるが、しかしながら、農業種に関しては限られている。例えば、アノテーション、可視化およびIntegrated Discovery (DAVID) は、主にヒトおよびヒトの疾患に焦点を合わせた任意の所与の遺伝子リストに基づく機能的アノテーションおよび分析の包括的なプラットフォームを提供する。The Gene Ontology enRIchment anaLysis and visuaLizAtion tool (GORILLA)をモデル種エンリッチメント解析に使用されている。本著者らの研究室は農業コミュニティのために2つのGOベースのツール、easyGO(ref.2)とそれに続くagriGO(ref.8)を開発し、それらを継続的に維持してきた。ハイスループット技術の急速な発展に伴い、これらのWebサーバーは農業分野における膨大な量のシーケンスデータを含むことができない(ref.9–14)。
さらに、GOデータベースは月ごとにいくつかのバージョンでリリースされているが、用語の定義や用語間の親子関係はさまざまな情報源からの情報の統合に頼って変化している(ref.15)。多くの情報源や方法が、不規則なアノテーションレベルとgene identities (IDs) を引き起こしている(ref.15–19)。エビデンスコードおよび/または比較ゲノミクスに基づいて比較的より包括的で正確なバージョンが作成されており、かなりの誤検出は減少してきている(ref.20)。例えば、イネの遺伝子または遺伝子産物のGOアノテーションには、TIGR(ref.21)、Gramene release 50(ref.22)、Phytozome v11(ref.18)などのバージョン5.0、6.1、および7.0が含まれている。したがって、絶えず拡張するknowledgeのもとで、GOアノテーション、および対応するGO termを増やす必要がある。
Google Scholarは、agriGOがオンラインになってから2017年1月までに982回参照され、8万件を超える分析リクエストを出していることを示した。同時に、ユーザーは信頼できる背景を持つ正確な分析結果に対する一連の要求を提示した。増え続ける配列種を満たし、最新のGOの背景(ref.15)を維持し、ユーザーの要件を満たすために、agriGOに新しい機能と優れたサポートを追加した。主なアップデートは、より多くの種とデータ型をサポートし、新しい視覚化ツールを追加することである。
AgriGO v2.0は主にagriGO v2.0が提供するバックグラウンドおよびカスタム分析を処理するための分析ツールを含んでいる。検索、singular enrichment analysis (SEA)、parametric analysis of gene set enrichment (PAGE)、SEACOMPARE、Batch SEA、DAGドロワー散布図(論文図1)。前の4つはagriGOから派生し、幅広い種とデータ型で更新されたが、DAGと散布図は視覚化のために新しく開発された。 SEAとBatch SEAの違いは、後者が1つのリファレンスに基づいて複数の入力データセットに同時に取り組むことができることである(論文図2)。 SEACOMPAREは、Batch SEAの結果からのfalse discovery rates (FDRs) に基づき、重要なGO termsを比較するのにも便利である(論文図3)。

The analysis framework for agriGO v2.0. 論文より転載
マニュアル
http://systemsbiology.cau.edu.cn/agriGOv2/manual.php
FAQ
http://systemsbiology.cau.edu.cn/agriGOv2/FAQ.php
使い方
http://systemsbiology.cau.edu.cn/agriGOv2/ にアクセスする。

*私だけかもしれませんが、夕方以降はログインできないことが多いです。
--リストのsubmit--
step1
上のAnalysisタブから生物種を選ぶ。

Otherにかなり登録されています。忘れず確認してください。
step2
speciesを選択したら、初めに方法を選ぶ。 singular enrichment analysis (SEA)分析(SEAは各termを独立して評価する。 term間の階層的関係は無視される、リストが多いと重い)、gene set enrichment analysis (GSEA)(PAGEはよりたくさんの遺伝子/プローブリストを扱え、長いリストの時に使う。fold change情報が必要)等を選べる。SEAはリストのみで実行できるが、PAGEは SEAとは異なり発現レベルを考慮に入れている。よって発現データがない場合はSEAを選択する。
SEACOMPAREはSEAの結果を比較するために使う(後述)。REViGOはGO termの長いリストを入力とし、冗長なGO termを削除し、要約GOと図を出力する。

step3
方法を決めたら、用意したIDリストを入力する。先ほど述べたように、PAGE分析は発現レベルを考慮に入れるので、最低2行、すなわち シーケンスID(1行目)、fold change(2行目以降)を入力する必要がある。fold changeはlog2 FCがより望ましい。

time seriesなど複数条件ある場合は、3行目以降に順番に指定していく。詳細は各生物のexampleデータの例を参照。
step4
パラメータ設定 (option)

SEA分析では、超幾何学的検定、カイ二乗検定およびフィッシャー検定の3つの検定法から選択する。事前計算のバックグラウンドと比較する場合、または参照リストのサブセットの場合は、超幾何またはフィッシャー検定を選ぶ。クエリリスト番号と参照リストの両方が非常に小さい場合は、フィッシャーテストを選ぶことが推奨されている。一方、 入力と参照のリストとの交差がほとんどない場合はカイ2乗検定が推奨されている。その下のMulti test adjustment methodは、 7つの調整方法:Yekutieli(FDR依存)、Bonferroni、Hochberg、Hochberg(FDR)、Hommel、Holm、False Discovery Rateからマルチテスト調整方法を選択できる。調整テストをオフにして使用することもできる(falseが増える)。 そのほか、有意水準のカットオフも指定できるようになっている。 Minimum number of mapping entriesは、指定以下のエントリ数しか現れないGOを除く設定になる。
step5
submitを押して実行する。
--出力--
1、direct acyclic graph
SEAの出力例

3つのGOカテゴリーそれぞれについて別々のグラフとして生成される。出力フォーマット、グラフのランク方向(上 <=> 下)、フォントサイズを選んで画像を描画する。
PNGでCellular Componentを描画してみた。

画像に表現されているのは統計的に有意なtermのGO階層ノードになる。 これらのノードは、対応する特定の色に関連付けられている10のレベルに分類される。設定した多重検定調整のp値が小さいほど統計的に有意で、ノードの色は濃くなる(赤に近づく)。 統計的に重要なtermのボックス内には、GO term、p値、GOの説明、クエリリスト内のGOと背景をマッピングするアイテム番号、クエリリストと背景の総数が含まれるが、p値がユーザーが設定カットオフよりも高い場合、ボックス内にはGO情報のみが表示される。 PNG、JPG、またはGIF形式で描画した時は、ブロックをクリックすることで termの詳細へのリンクを開くことができる。
PAGEの出力例
PAGE分析の結果はSEA分析とかなり類似している。PAGEツールは一度に複数の行を分析でき、各行のGO termが計算される。 各行の有意なGO termの数が要約部分に表示される。

継時的な発現解析で複数カラム入力しているため、カラムごとにまとめられる。
Detailsをクリックすると有意なGO termをまとめたページにジャンプする。下の図は0-24hまであるが、term数は、クリックしたDetailsリンク(ここでは24hをクリックしており、下の図の右端の24hで有意なGO termのみが並ぶ)のGOリストのみになる。 CMという名前のカラムで使用されている色は、グラフィック表示で使用されている色と同じになる。赤のカラーシステムはup regulation、青色はdown regulationを意味する。 各ブロックはその行のGO termのZスコアを表す。

ここで特定のGO termにチェックをつけ、上のボタンをクリックして新しい画像を生成できる。しかしテスト時は動作しなかった(Firefox推奨だがテストはSafariで行った)。
わかりにくいが、表の数値部分をクリゥクすればGO term詳細を確認できる。

SEAと同様に画像も出力できる。発現情報を加味して色がつけられる。

2、チャートはテスト時動作しなかった。
Batch Singular Enrichment Analysis (SEA)の使い方はweb manualを参照してください。
http://systemsbiology.cau.edu.cn/agriGOv2/manual.php
引用
agriGO v2.0: a GO analysis toolkit for the agricultural community, 2017 update
Tian Tian, Yue Liu, Hengyu Yan, Qi You, Xin Yi, Zhou Du, Wenying Xu, Zhen Su
Nucleic Acids Res. 2017 Jul 3; 45(Web Server issue): W122–W129.
agriGO: a GO analysis toolkit for the agricultural community
Zhou Du, Xin Zhou, Yi Ling, Zhenhai Zhang, and Zhen Su
Nucleic Acids Res. 2010 Jul;38(Web Server issue):W64-70
補足
非モデル生物でもGO termをアサインすれば使うことはできる。
BLAST4IDを選択する。
BLASTPならタンパク質配列をアップロードする。ファイルサイズ上限が4MBと小さいので時には複数回アップする必要がある。混雑していることが多いので、結果が得られるまでかなりの時間がかかる可能性がある。
外部リソースを使った場合、例えばegg NOG mapperを使うとGO termやKO termを各遺伝子やタンパク質にアサインできる。egg NOG mapperのバージョンによって変わる可能性はあるが、下のアノテーション結果では10列目がGO term。
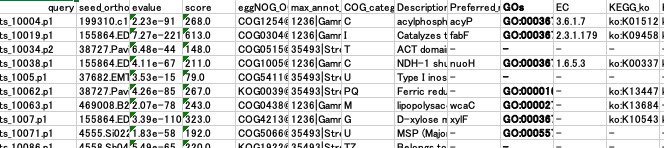
1つの遺伝子に複数のGO termがついている事が多いので、1行1語彙になおす。ここではRを使う。
#1 - egg NOG mapper.csvのコメント行を消す。
grep -v "#" eggnog.csv > filtered.csv
#2 - R
> library(data.table) #conda
> eggnog <- read.csv("filtered.csv", header = TRUE, sep = ",")
> go <- eggnog[c(1,10)] #1列目が遺伝子、10列目がGO
> go <- data.table(go) #データフレームに変換
#分割する。入力CSVのヘッダがgene.idとGOsで、複数GO termある時にカンマ","で各語彙が区切られているなら以下のように打つ。
> go_trans <- go[, list(GOs = unlist(strsplit(GOs , ","))), by = gene.id]
> write.table(go_trans, "GO_background.tsv", quote=F, col.names=T, append=F, sep="\t")
#カレントパスに保存
DEGsのリストも同様に処理する。agriGOでカスタムを選択し(以下はSEAの例)、クエリ(DEGs)とバックグラウンドどちらも遺伝子名<tab>GOterm(写真のように)入力する。バックグラウンドはその生物の全遺伝子のGOリストか、その組織で発現している全ての遺伝子のGOリストとする。de nono transcriptomeならアセンブルした全ての転写産物のGO termをバックグラウンドとする。

テキストをアップロードしてもよい。