ハイスループットシーケンス(HTS)データからの情報マイニング用にさまざまなソフトウェアまたはパイプラインが開発されているが、それらのほとんどは、ほとんどの生物学者が馴染みのないプログラミングおよびコマンドライン環境に依存している。 ユーザーフレンドリーなインターフェースを備えたバイオインフォマティクスツールは、ウェットラボの生物学者に好まれている。 ここでは、さまざまなHTSデータ処理ツールと使いやすいインターフェイスを統合した生物学者向けのツールキットであるTBtoolsについて説明する。 バルクシーケンス抽出、gene set functional enrichment、ベン図など、HTSデータで作業する多くのシンプルで日常的で精巧なタスクを容易にする多数のコレクションが含まれている。TBtoolsは、JRE1.6および github.com/CJ-Chen/TBtoolsから無料で入手できる。 その開発以来、多くの研究者によって使用されてきた。 ウェットラボの生物学者があらゆる種類のハイスループットデータを扱うのに役立つツールキットである。
Manualより
TBtoolsは何ができるの?
TBtoolsは、"Tools for Biologist "の略で、ツールセットであり、プロジェクトでもあります。最初から、私はただツールセットを開発したいと思っていました。それは、私自身のためのコマンドラインモードと、私の同僚のためのグラフィック・ユーザー・インターフェースモードです。しかし、何人かの友人がこのworksを見て、TBtoolsは他の方にも役立つだろうと言いました。そこで、私はそれをウェブに掲載しました。その結果、より多くの友人が私に機能要求を送り、TBtoolsにますます多くの機能が追加されました。現在(2017/06/29)まで、TBtoolsには以下のような機能が含まれています。
1、シーケンスツール
- Amazing Fasta Extractor Fastaファイルの変換、Fastaインデックスの作成、Fastaレコードの抽出を短時間で行うことができます。
- Fasta Stater Fastaファイルの統計を所得します。全Fastaレコードのサマリー情報(レコード数、トータル長、N50、GCコンテンツなど)と、各Fastaレコードの配列特徴(簡略化されたID、長さ、GCコンテンツなど)を含むファイルを生成することができます。
- Fasta Extractor Fastaファイルから直接Fastaレコードを抽出しますが、Amazing Fasta Extractorよりも遅いかもしれません。この機能は将来的に減少または更新される予定です... 代わりにAmazing Fasta Extractorを使うことをお勧めします。
- Fasta Subseq Fasta Extractorと似ていますが、特定のFastaレコードのSequence Regionを抽出するために開発された機能です... この機能は今後減少または更新される予定です... Amazing Fasta Extractorの使用をお勧めします。
- Fasta Merge and Split 複数のFastaファイルを1つのFastaファイルに結合したり、1つのFastaファイルを複数のFastaファイルに分割したりします。
- Sequence Manipulator シーケンスのトランスフォーマット、リバース、コンプリメンテーション
- NCBI Seq Downloader NCBIからGIやAccession Numberのリストに従ってSequncesを一括でダウンロードします。
- Get Completes ORF(Open Reading Frame) 入力配列から完全なORFを予測します。現在のところ、この機能は完全なORFと古典的なコドン使用表のみを検出します。つまり、完全なORFとは、ATGから始まり、TGA、TAA、TAGで終わる配列領域のみを指します。
- Check Primers(Simple PCR) トランスクリプトームなどの特定のシーケンスデータベースに入力されたプライマーの仕様を簡単に統計するために、プライマーの配列位置を直接確認します。
- GFF/GTF Sequence Extractor 遺伝子構造アノテーションファイル(.gff/.gtf)に基づいて、ゲノムから塩基配列を抽出します。
2、BLASTツール
- Remote Blast(No Need for Preinstalled Blast) JRE1.6がSSLプロトコルをサポートしておらず、このためにサードパーティのライブラリを追加したくないため、この機能は現在安定していません。後で更新されます。
- Auto Blast Several Sequences To a Big File (よく使う機能) 環境内でBlastを起動して、複数のFasta形式の大きな配列ファイルを比較します。
- Auto Blast Two Sequence Sets 上記と同様です。
- Auto Blast Two Sequence Sets -Big File 上記のような機能です。この3つの機能は、機能の中で1つに統合される予定です。
- Blast Several Seq To FastQ 入力されたFastQファイルからBLASTデータベースを構築し、複数の配列のBLASTサーチを行います。
- Reciprocal Blast fasta形式の2つの入力ファイル間で相互にBLASTを行います。
- Blast XML Alignment Shower BLAST結果のアラインメントグラフを作成し、クエリ配列や対象配列のカバレッジの確認に使用できます。
- Blast XML Dotpot BLAST結果のドットプロットグラフを作成します。
- Blast Pileup Grapher NCBI blast web serveive result pageに似た、BLAST結果のpileup graphを作成します。
- TransFormat Blast.xml to TBtools.table TBtoolsは、BLAST結果を保存して記述するために、タブ形式のフォーマットを定義しています。このテーブルは、weighted-Covのようないくつかの有用な静的情報を含んでいます。
- TransFormat Blast.xml to Blast Table blast.xmlファイルを、デフォルトのBLAST+ outfmt-6と同じタブ区切りのファイルに変換します。
- e-GenomeWalkiing or e-Race オーバーラップするリードをFISHして、長い配列にまとめるシークエンスを使用します。この機能は、リシーケンスデータのGenome-walkingや、RNAシーケンスデータの5'RACEや3'RACEをsilcoで行う際に便利です。
3、GO・KEGGのツール
- GO Annotation 遺伝子関連のアノテーションを行います。単純にNCBI, Uniprot, TrembleのGI,Accession NumberをGO IDにマッピングするだけのIDマッピングツールです。
- GO Enrichment 超幾何学的分布に基づいて、 gene-ontology term enrichment analysisを行います。
- GO Level Counter 特定のGOレベルの遺伝子数をカウントし、統計表と任意のグラフを出力します。
- GO Level Compare 特定のGOレベルでの2つのGOアノテーション結果を比較し、グラフを出力します。この機能は現在うまく動作していません。修正します。
- GO Term Parser GOアノテーションを解析して、Gene2GOやGO2geneのマッピング情報を得ることができます。
- Prepare GO Annotation for BiNGO in cytoscape 多くの遺伝子アノテーションをBiNGO用のフォーマットファイルに変換します。
- KEGG Enrichment Analysis 超幾何学的な分布に基づいたKEGGパスウェイ解析を行います。
- KEGG Pathwat Map Drawer KEGG パスウェイマップ上の遺伝子をハイライト表示します。
4、その他のツール
- Color Picker カラーコードを自由に選び、必要に応じてカラーコードグラフを保存することができます。
- Table ID Manipulator 表の行を抽出、フィルタリング、ランク付けすることができます。
- Table Column Manipulator ランク付けしたり、選択した列だけを残したりします。抽出・フィルタ機能が追加される予定です。
- Big Text Viewer 非常に大きなサイズのテキストファイルを短時間で表示します。
- Big Table Viewer 大きなサイズのテーブルファイルを簡単に見ることができます。
- Text Block Extractor テキストブロックを、特定のIDリストと記録された個別の文字列で抽出します。この機能は、シンテニーブロックを抽出するために開発されました。
- Expression Shower 1つの遺伝子または複数の遺伝子の発現傾向を可視化します。
- Expression Calculator gene.countsとgene.lenに基づいて、発現量(RPKMまたはTPM値)を計算する。
- Wonderful Venn (Up to Six Sets) Venn解析をインタラクティブに行うことができます。
- Map Genes On Genome From Sequence Files Blastを用いて入力遺伝子のゲノム上の位置領域を取得し、グラフを出力します。
- Map Genes On Genome From Position Info File 入力された遺伝子とゲノム情報をもとに、ゲノムファイル上に遺伝子を描画します。
- Dual Synteny Plot from MCScanX output MCScanXの結果をインタラクティブに表示することができます。
- Domain/Motif Pattern Drawers MEME suite、NCBI Batch-CD search、pfam-search、GFF/GTFの結果を表示します。
より最新のドキュメントはどうやら中国語だけで書かれており、見つけることが出来なかった。
インストール
TBtools is a platform-independent software that can be run under all operating systems with Java Runtime Environment 1.6 or newer. I
リリースより各OS向けにビルドされた.jarファイルやインストーラー(.exe, .dmg)をダウンロードできる。
指示に従ってインストールする(windowsの場合)。
立ち上げる。

起動したところ。
実行方法
機能が膨大なので、基本的なインターフェイスと操作メニューだけ確認していきます。
1、シーケンスツール
様々な機能を持つ。

シーケンスツールには、配列操作に関するツール、NCBIからの配列ダウンロード、primer配列のチェック、GFF, GTF関連のツールがある。
Fasta statsを使ってみる。
Sequence Toolkit => Fasta Statsを選択
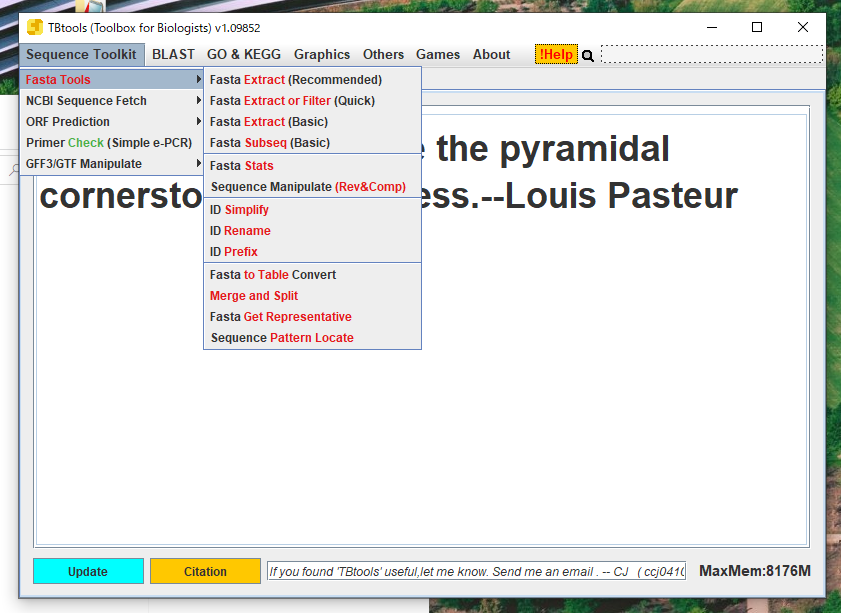
新しいタブが表示された。TBtoolsはこのようにタブを切り替えて各ツールを管理する。
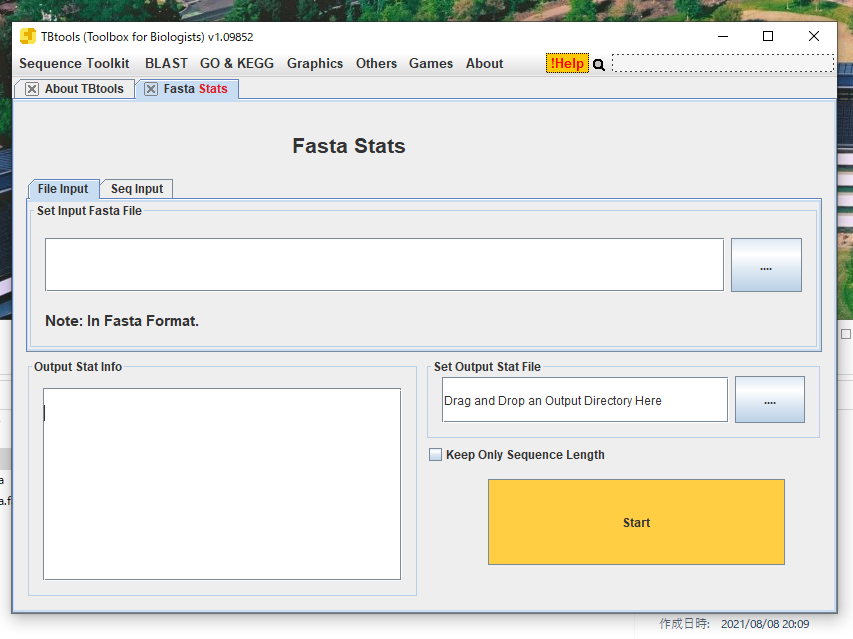
塩基配列かアミノ酸配列のfasta配列をドラッグして読み込ませる。読み込まれると別のウィンドウが出現して配列を確認できる。more, lessコマンドのようにページ送りできる。
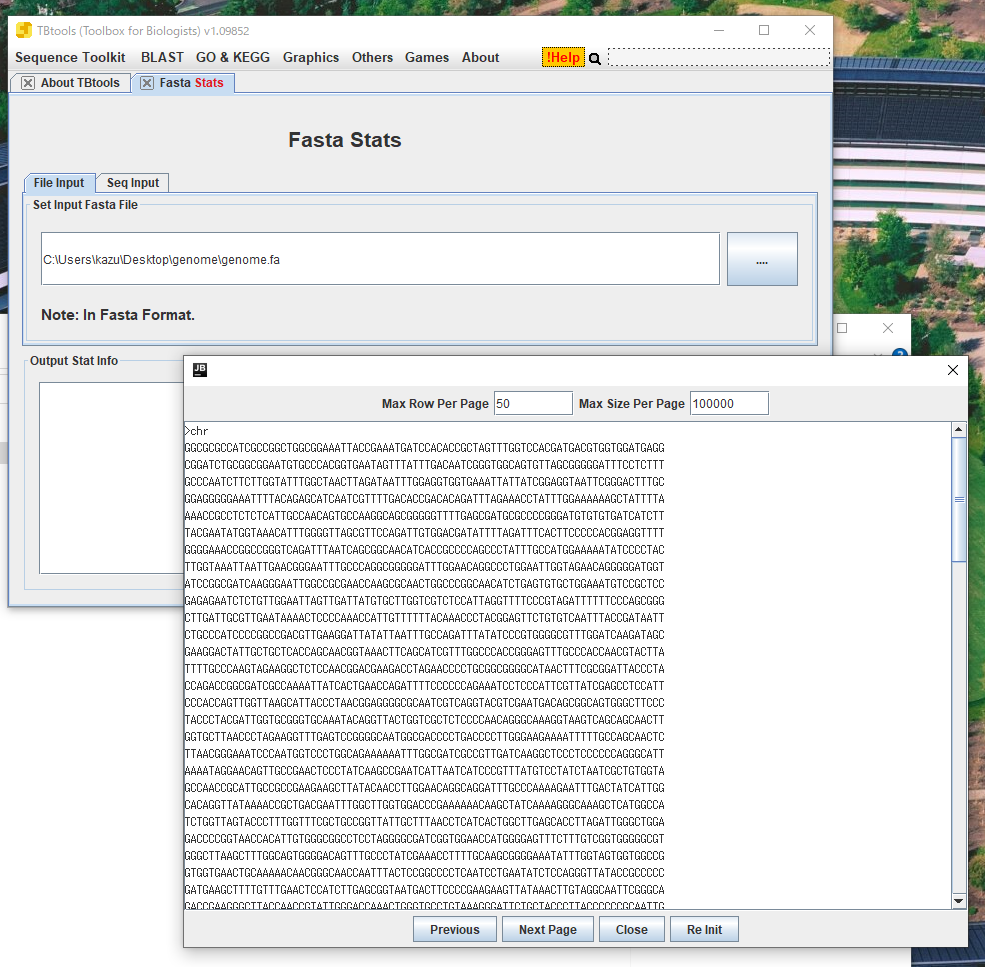
確認したら閉じる。
Startボタンをクリックすると配列の基本的な統計がプリントされる。
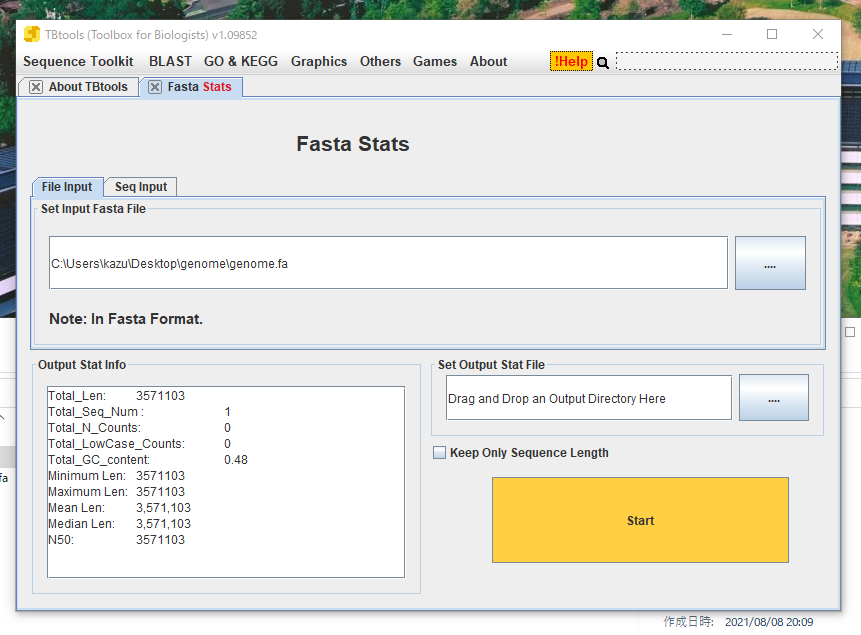
出力ディレクトリを指定すると、.xls形式で統計結果が保存される。

2、BLASTツール
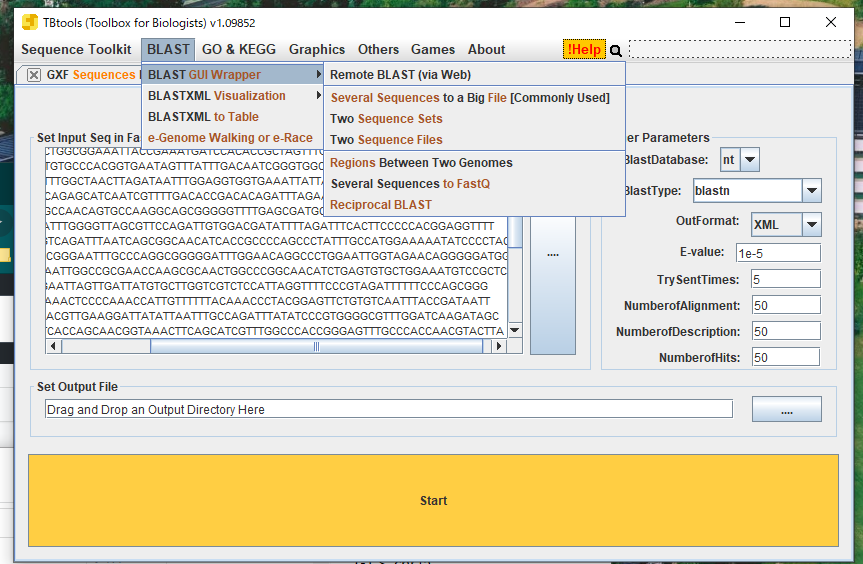
BLAST検索して様々な方法、例えばdot plotなどで結果を表示できる。

3、GO・KEGGのツール

4、Graphicsツール (2017年のマニュアルでは触れられていない)

Heatmap

Sequence logo

Basic PCA

Volcano plot
Gene structure view

Advanced circos

Synteny visualization

Others

実際の使い方については2020年に出版された論文でも説明されています。アクセスして確認して下さい。
引用
TBtools, a Toolkit for Biologists integrating various HTS-data handling tools with a user-friendly interface
Chengjie Chen, Rui Xia, Hao Chen, Yehua He
bioRxiv preprint first posted online Mar. 27, 2018
TBtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data
Chengjie Chen, Hao Chen, Yi Zhang, Hannah R Thomas, Margaret H Frank, Yehua He, Rui Xia
Mol Plant. 2020 Aug 3;13(8):1194-1202