従来の形態学的同定による水生侵入種の検出は、しばしば時間がかかり、高度な分類学的専門知識を必要とし、緩和対応の遅れにつながる可能性がある。これらの障害を克服するために、Illumina ベースのシーケンシング技術を用いた複数種の環境 DNA (eDNA) 検出アプローチが使用されてきたが、サンプル処理には時間がかかることが多い。最近では、ポータブルなナノポアシーケンシング技術が利用できるようになり、侵略的な種の分子検出をより広く利用できるようになり、サンプルのターンアラウンドタイムを大幅に短縮できる可能性を秘めている。しかし、ナノポアシーケンシングされたリードは、Illuminaプラットフォームで作成されたものよりもはるかに高いエラー率を持っており、これまでのところ、この技術の採用を妨げてきた。著者らは、侵襲種を検出するためのナノポア配列決定の信頼性を高めるための詳細な実験プロトコルとバイオインフォマティクスツールを提供し、侵襲的な二枚貝を使用してそのアプリケーションをテストした。イタリアとポルトガルで、既存の二枚貝の発生と豊富さのデータがあり、対照的な二枚貝の群集がある場所から水をサンプリングした。3.5日でeDNAの抽出、増幅、シークエンシングを行った。処理されたリードの大部分は、参照配列と99%以上同一であった。発生が知られているもの以外の分類群は検出されなかった。いくつかのサイトでいくつかの種が検出されなかったのは、その存在量が少ないことが知られているためと説明できるかもしれない。これは、eDNAサンプルから水生侵入種を検出するためにMinIONを使用した最初の報告である。このアプローチは、生物多様性評価、生態系の健全性評価、食事研究など、他のメタバーコーディングのアプリケーションにも簡単に適応できる。
インストール
ubuntu18.04LTSでmambaを使って導入した。
mamba create -n metabinkit -c bioconda -c conda-forge metabinkit python=3.8 -y
conda activate metabinkit
> metabin -h
$ metabin -h
Usage: metabin [options]
Options:
-i FILENAME, --input=FILENAME
TSV file name
-o FILENAME, --out=FILENAME
output file prefix
-S DOUBLE, --Species=DOUBLE
species %id threshold [default= 99]
-G DOUBLE, --Genus=DOUBLE
genus %id threshold [default= 97]
-F DOUBLE, --Family=DOUBLE
family %id threshold [default= 95]
-A DOUBLE, --AboveF=DOUBLE
above family %id threshold [default= 90]
-D FOLDER, --db=FOLDER
directory containing the taxonomy db (nodes.dmp and names.dmp) [default= /home/kazu/miniconda3/envs/metabinkit/bin/../db/]
--SpeciesNegFilter=FILENAME
negative filter (file with one word per line) [default= NULL]
--SpeciesBL=FILENAME
species blacklist (file with one taxid per line) [default= NULL]
--GenusBL=FILENAME
genera blacklist (file with one taxid per line) [default= NULL]
--FamilyBL=FILENAME
families blacklist (file with one taxid per line) [default= NULL]
--FilterFile=FILENAME
file name with the entries from the input to exclude (on entry per line) [default= NULL]
--FilterCol=COLUMN NAME
Column name to look for the values found the the file provided in the --Filter parameter [default= sseqid]
--rm_predicted=COLNAME
Where to look (column name) for in-silico 'predicted' entries (XM_,XR_, and XP_). If no column is given then the filter is not applied. [default= NULL]
--TopSpecies=INTEGER
[default= 100]
--TopGenus=INTEGER
[default= 100]
--TopFamily=INTEGER
[default= 100]
--TopAF=INTEGER
above family? [default= 100]
-v, --version
print version and exit
-q, --quiet
enable quiet mode (less messages are printed to stdout)
--no_mbk
Do not use mbk: codes in the output file to explain why a sequence was not binned at a given level (NA is used throughout)
--sp_discard_sp
Discard species with sp. in the name
--sp_discard_mt2w
Discard species with more than two words
--sp_discard_num
Discard species with numbers
-M, --minimal_cols
Include only the seqid and lineage information in the output table [FALSE]
-h, --help
Show this help message and exit
> metabinkit_blast -h
テストラン
1、データの準備
metabinkitはタブ区切りのTSVファイルを要求する。このTSVファイルには、qseqid, pident, taxids の 3 列が必要。オプションで K, P, C, O, F, G, S の 7 つの列も追加できる。
- qseqid:クエリ配列のID
- pident: アラインメントの同一性の割合
- taxids: NCBI taxid:データベース対象配列のNCBI taxid
- (optional) K,P,C,O,F,G,S: データベース対象配列の kingdom, pylum, class, order, family, genus, species
テスト用のTSVファイルを確認する。
git clone https://github.com/envmetagen/metabinkit.git
head metabinkit/tests/test_files/in0.blast.short.tsv

デフォルトでは、BLASTパラメータをチューニングした「徹底的な」BLASTを実行するためのBLASTのラッパースクリプトも用意されている。感度が高いため、デフォルト設定より多くのCPU時間を必要とする。
metabinkit_blast -f input.fasta -D reference_DB -o out.tsv
2、metabinのラン
TSVファイルが用意できたら実行する。ここではテストファイルを使う。
metabin -i metabinkit/tests/test_files/in0.blast.short.tsv -o out0.short.bins
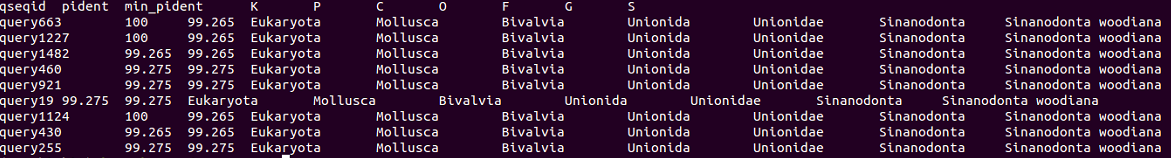
出力

head out0.short.bins.tsv
引用
Speeding up the detection of invasive aquatic species using environmental DNA and nanopore sequencing
Bastian Egeter, Joana Veríssimo, Manuel Lopes-Lima, Cátia Chaves, Joana Pinto, Nicoletta Riccardi, Pedro Beja, Nuno A. Fonseca
bioRxiv, Posted June 11, 2020