2024/02/27 追記
Horizontal gene transfer(HGT)は原核生物のゲノムの変動性の主な原因である。ゲノム可塑性領域(Region of genome plasticity: RGP)とは、非常に可変性の高いゲノム領域に位置する遺伝子の集合のことである。その多くはHGTから発生し、 genomic islands(GI)に対応している。これらの領域を種レベルで研究することは、ゲノムデータの氾濫に伴い、ますます困難になってきている。現在までのところ、数百のゲノムを用いてGIを同定し、その多様性を探る方法はない。
本研究では、ある種の全ゲノムから作成したパンゲノムグラフを用いてRGPを予測するpanRGP法を提案する。これにより、RGPの多様性にアクセスし、挿入のスポットを予測するために、何千ものゲノムを研究することができる。他のGI検出ツールと基準データセットとのベンチマークを行ったところ、最高の予測結果が得られた。さらに、大腸菌の挿入スポットとしてよく研究されているleuX tRNAホットスポットの境界を再定義することで、メタゲノムをアセンブルしたゲノム上でのpanRPGの使用を説明した。
本研究で紹介した手法は、以下のソフトウェアから入手できる: https://github.com/labgem/PPanGGOLiN。ベンチマークメトリクスを計算するための詳細な結果とスクリプトは https://github.com/axbazin/panrgp_supdata から入手できる。
インストール
ubuntu18.04LTSでテストした。レポジトリではmambaによる導入が推奨されているが、ここではcondaコマンドでcondaの仮想環境に導入した。
本体 Github
#bioconda (link)
mamba create -n ppanggolin-env -y
conda activate ppanggolin-env
mamba install -c bioconda ppanggolin -y
> ppanggolin rgp -h
$ ppanggolin rgp -h
usage: ppanggolin rgp [-h] [--persistent_penalty PERSISTENT_PENALTY]
[--variable_gain VARIABLE_GAIN] [--min_score MIN_SCORE]
[--min_length MIN_LENGTH] [--dup_margin DUP_MARGIN] -p
PANGENOME [--tmpdir TMPDIR] [--verbose {0,1,2}]
[--log LOG] [-c CPU] [-f]
Optional arguments:
--persistent_penalty PERSISTENT_PENALTY
Penalty score to apply to persistent genes (default:
3)
--variable_gain VARIABLE_GAIN
Gain score to apply to variable genes (default: 1)
--min_score MIN_SCORE
Minimal score wanted for considering a region as being
a RGP (default: 4)
--min_length MIN_LENGTH
Minimum length (bp) of a region to be considered a RGP
(default: 3000)
Minimum ratio of organisms where the family is present
in which the family must have multiple genes for it to
be considered 'duplicated' (default: 0.05)
Required arguments:
One of the following arguments is required :
-p PANGENOME, --pangenome PANGENOME
The pangenome .h5 file (default: None)
Common arguments:
-h, --help show this help message and exit
--tmpdir TMPDIR directory for storing temporary files (default: /tmp)
--verbose {0,1,2} Indicate verbose level (0 for warning and errors only,
1 for info, 2 for debug) (default: 1)
--log LOG log output file (default: stdout)
-c CPU, --cpu CPU Number of available cpus (default: 1)
-f, --force Force writing in output directory and in pangenome
output file. (default: False)
> ppanggolin panrgp -h
$ ppanggolin panrgp -h
usage: ppanggolin panrgp [-h] [--fasta FASTA] [--anno ANNO]
[--clusters CLUSTERS] [-o OUTPUT]
[--basename BASENAME] [--rarefaction]
[-K NB_OF_PARTITIONS] [--interest INTEREST]
[--no_defrag] [--tmpdir TMPDIR] [--verbose {0,1,2}]
[--log LOG] [-c CPU] [-f]
Input arguments:
The possible input arguments :
--fasta FASTA A tab-separated file listing the organism names, and
the fasta filepath of its genomic sequence(s) (the
fastas can be compressed). One line per organism. This
option can be used alone. (default: None)
--anno ANNO A tab-separated file listing the organism names, and
the gff filepath of its annotations (the gffs can be
compressed). One line per organism. This option can be
used alone IF the fasta sequences are in the gff
files, otherwise --fasta needs to be used. (default:
None)
--clusters CLUSTERS a tab-separated file listing the cluster names, the
gene IDs, and optionnally whether they are a fragment
or not. (default: None)
Optional arguments:
-o OUTPUT, --output OUTPUT
Output directory (default: ppanggolin_output_DATE2021-
01-05_HOUR12.54.09_PID2944099)
--basename BASENAME basename for the output file (default: pangenome)
--rarefaction Use to compute the rarefaction curves (WARNING: can be
time consumming) (default: False)
-K NB_OF_PARTITIONS, --nb_of_partitions NB_OF_PARTITIONS
Number of partitions to use. Must be at least 3. If
under 3, it will be detected automatically. (default:
-1)
--interest INTEREST Comma separated list of elements to flag when drawing
and writing hotspots (default: )
--no_defrag DO NOT Realign gene families to link fragments with
their non-fragmented gene family. (default: False)
Common arguments:
-h, --help show this help message and exit
--tmpdir TMPDIR directory for storing temporary files (default: /tmp)
--verbose {0,1,2} Indicate verbose level (0 for warning and errors only,
1 for info, 2 for debug) (default: 1)
--log LOG log output file (default: stdout)
-c CPU, --cpu CPU Number of available cpus (default: 1)
-f, --force Force writing in output directory and in pangenome
output file. (default: False)
テストラン
ランするには生物名リストファイル(テストファイルは”ORGANISMS_FASTA_LIST”)を指定する。このリストファイルは、最初のカラムにゲノムファイル名(ユニークであること)(空白は不可)、タブ区切り、2 番目の列にそのFASTA ファイルへのパスを記載する。環状の配列が含まれている場合、タブ区切りで3列目以降にその識別子を記載する。
リストファイルを指定してラン
git clone https://github.com/labgem/PPanGGOLiN.git
cd PPanGGOLiN/testingDataset/
ppanggolin panrgp --fasta organisms.fasta.list--cpu 20 -o outdir
50個のChlamydia trachomatis genomeのランはたった数十分で終わった。
出力
partitions/
Ushaped_plot.html
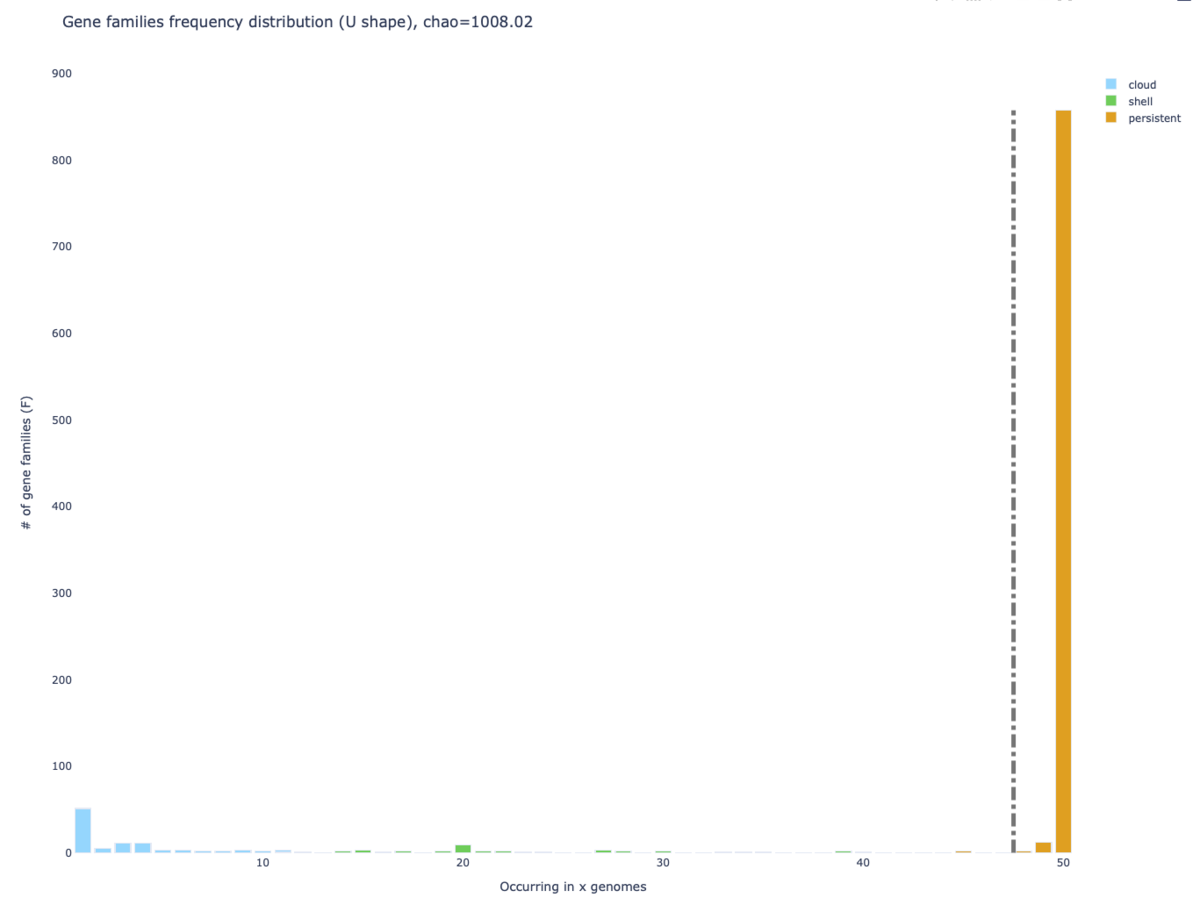
- ppanggolinの通常のパンゲノム解析ワークフローを実行後、ゲノムの可塑性領域( regions of genome plasticity)に関連するファイルが追加で書き込まれる。
- PanGGOLiNの統計的アプローチでロバストな結果を得るためには、ゲノムに変異を持つ15ゲノム以上を使用することが推奨されている。
出力内容の詳細はwikiのoutputを確認してください。
Outputs · labgem/PPanGGOLiN Wiki · GitHub
引用
panRGP: a pangenome-based method to predict genomic islands and explore their diversity
Adelme Bazin, Guillaume Gautreau, Claudine Médigue, David Vallenet, Alexandra Calteau Author Notes
Bioinformatics, Volume 36, Issue Supplement_2, December 2020
PPanGGOLiN の他のコマンドについては以前の投稿も参考にして下さい。