代表的なモデル生物6種(ヒト、マウス、ラット、ミミズバエ、線虫、出芽酵母)から得られた公開されているクロマチン免疫沈降シークエンス(ChIP-seq)とDNase-seqデータ(n>70,000)を完全に統合し、ChIP-Atlas(http://chip-atlas.org)と名付けられたデータマイニングプラットフォームを考案した。ChIP-Atlasは、GEO、ArrayExpress、DDBJ、ENCODE、Roadmap Epigenomics、科学文献から得られたデータを含むNCBI Sequence Read Archive(SRA)にアーカイブされているすべての公開ChIP-seqおよびDNase-seqデータのアライメントおよびピークコール結果を表示することができる。すべてのピークコールデータを統合して、特定のゲノム座位における複数のヒストン修飾と転写調節因子(TR)の結合部位を可視化する。統合されたデータは、TR-遺伝子とTR-TRの相互作用を示すために、さらに解析することができ、与えられた複数のゲノム座標や遺伝子名に対するタンパク質結合のエンリッチ度を調べることができる。ChIP-Atlasは、数千件に及ぶChIP-seq実験のデータマイニングにおいて、データ数や機能性の点で他のプラットフォームよりも優れており、遺伝子制御ネットワークやエピジェネティックなメカニズムについての洞察を提供している。
ChIP-Atlas: Peak Browser
Target Genes
Colocalization
Enrichment Analysis
2021 1/4
ChIP-Atlas のウェブサイトを色々と改善しました。データ検索やデータセットIDからデータの詳細を表示する機能が強化されてます。本年もどうぞご贔屓に! https://t.co/3ZjUsEvkmf
— 🤌🤌 (@iNut) 2021年1月4日
動画で説明されているので、ここでは流れだけ簡単に紹介します。
ChIP-Atlas にアクセスする。

主に4つの機能がある。各機能の下には解説した動画へのリンクも提供されている。
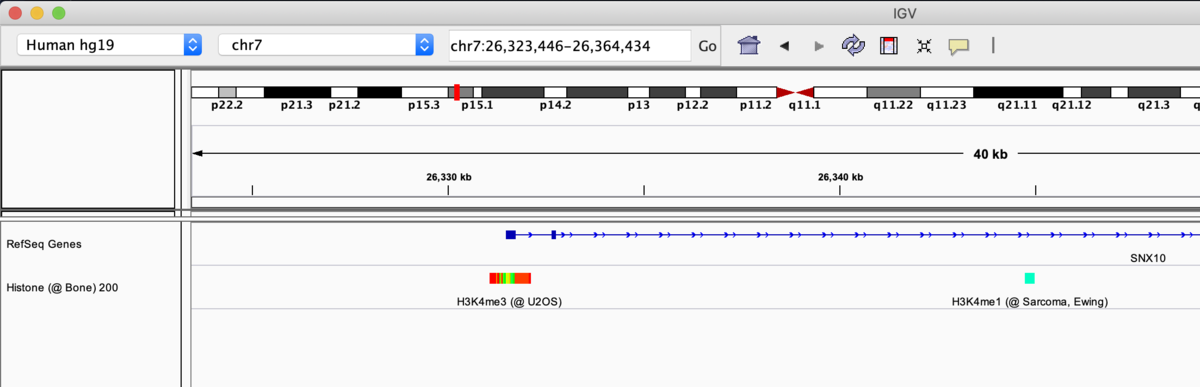
1、Peak Browser - IGVを用いて、ユーザーが指定したゲノムのタンパク質結合をグラフィカルに表示
最初にローカルマシンのIGVを立ち上げておく(持ってない人はダウンロードして下さい)。次に上のリファレンスゲノムのバージョンを指定する(デフォルトはhg19)。

それから抗原クラス、細胞のタイプ、閾値(MACS2で算出される統計的有意値の閾値。50を設定すると、Q値が1E-05未満のピークがIGVで表示される)を指定する。
 virew on IGVをクリックする。
virew on IGVをクリックする。
IGVで表示される。青は遺伝子。下の色付きボックスが転写因子。転写因子の色は結合の親和性の高さを表している。
2、Target Genes - 転写因子によって制御されるターゲット遺伝子を表示
リファレンスゲノムを指定し、転写因子名と転写開始点からの距離を指定する。

結果は縦軸がターゲット遺伝子、横軸が公共実験データ名の並びのヒートマップで視覚化される。色は親和性の高さを表す。左端にはaverageの列もある。
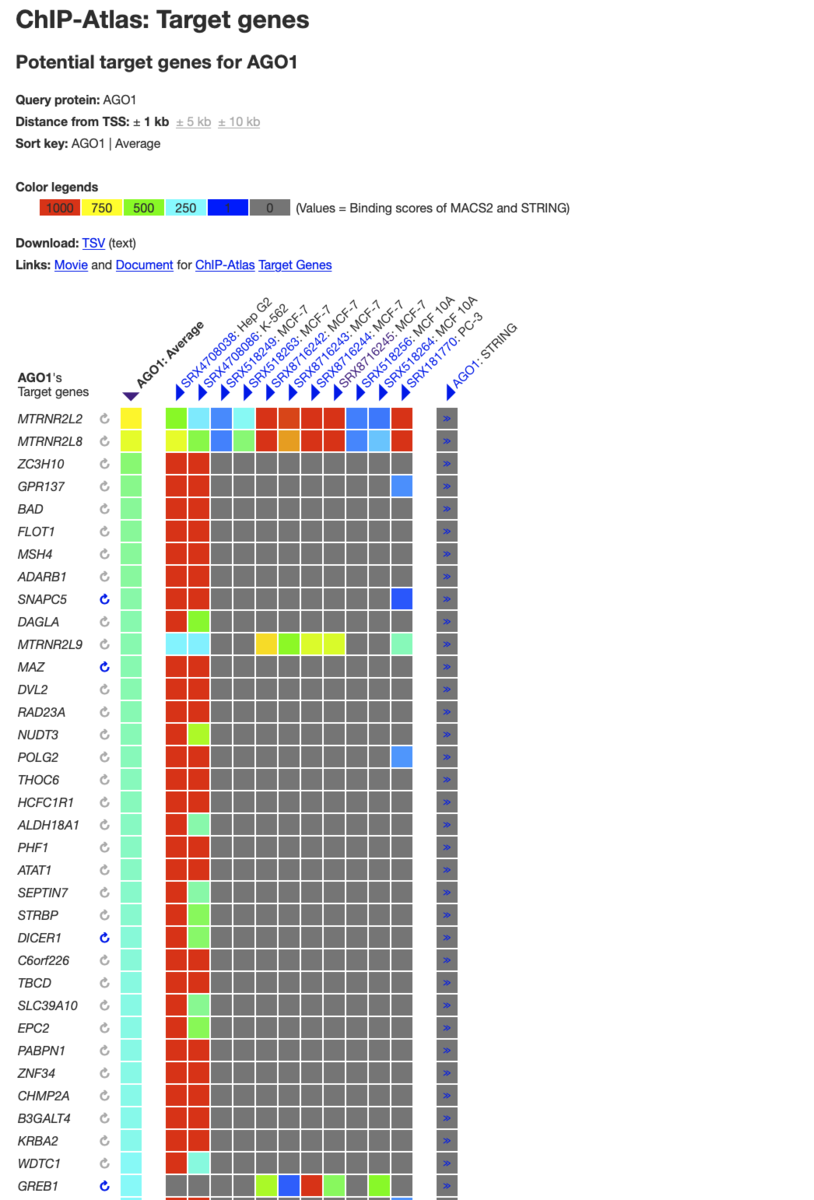
実験名を クリックすると詳細を確認できる。

3、Colocalization - 転写因子と共局在するパートナータンパク質を予測
転写因子を指定する。
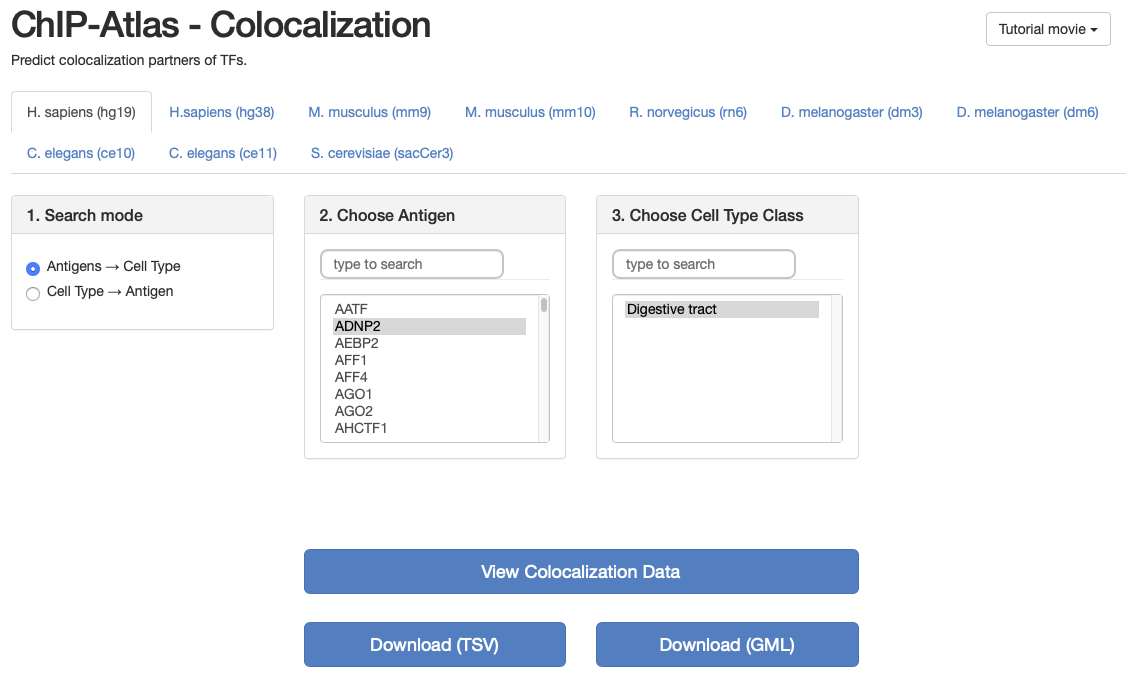
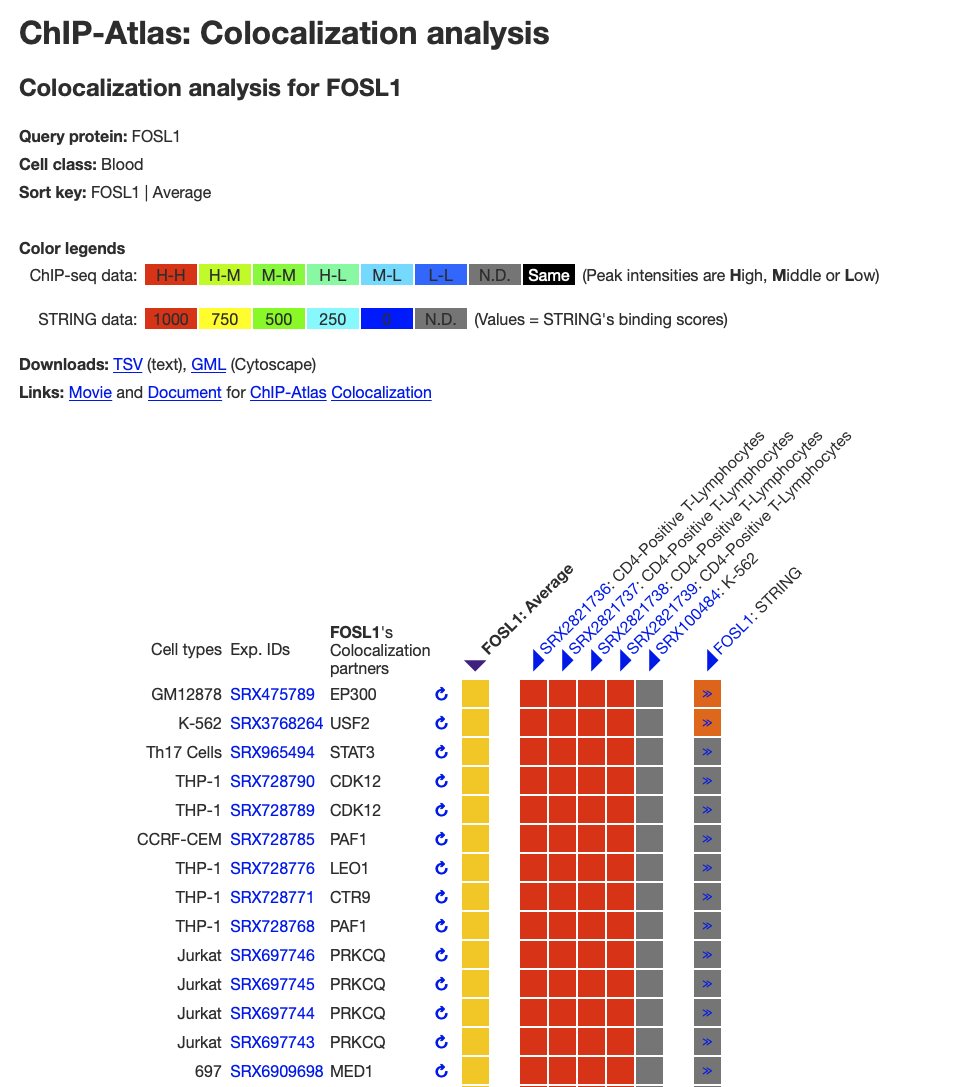
その転写因子と共局在する可能性のある転写因子が表示される。
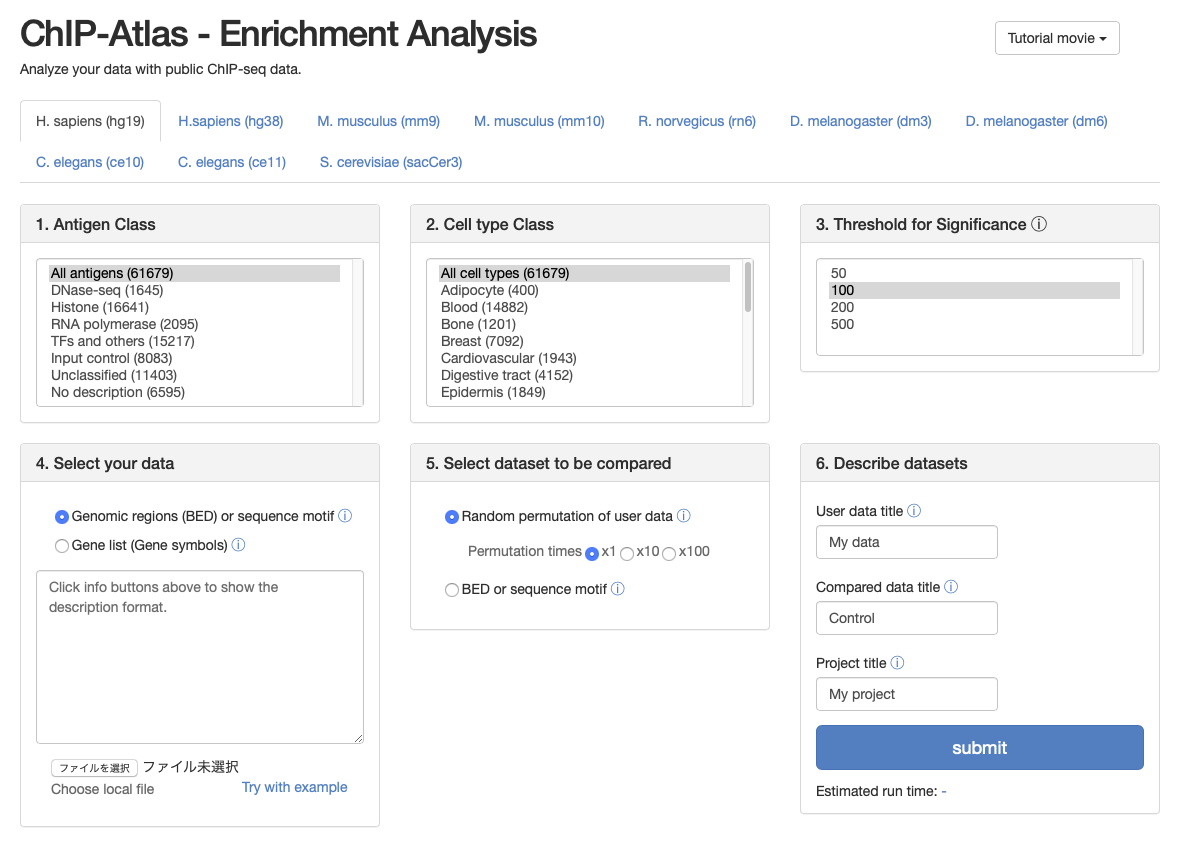
4、Enrichment Analysis - 与えられたゲノム座や遺伝子に結合したタンパク質を予測
引用
ChIP-Atlas: a data-mining suite powered by full integration of public ChIP-seq data
Shinya Oki, Tazro Ohta, Go Shioi , Hideki Hatanaka, Osamu Ogasawara, Yoshihiro Okuda, Hideya Kawaji, Ryo Nakaki, Jun Sese, Chikara Meno
EMBO Rep. 2018 Dec;19(12)
統合TVでも分かりやすく説明されています。
Peak Browserについて
Target Genesについて