細菌ゲノム疫学やアウトブレイク検出において、遺伝子ごとのアプローチがますます普及してきている。しかし、これらの方法論のためのスキーマ定義や対立遺伝子呼び出しのためのオープンソースのスケーラブルなソフトウェアが不足している。chewBBACAスイートは、新規の全ゲノムまたはコアゲノムの遺伝子ごとのタイピングスキーマの作成と評価、および関心のある細菌株におけるその後のアレルコールを支援するために設計されている。chewBBACAソフトウェアはPython 3.4以上を使用しており、ラップトップや高性能クラスタで動作するため、小規模な研究室から大規模なリファレンスセンターまで幅広く利用できる。ChewBBACA は https://github.com/B-UMMI/chewBBACA から入手可能である。
chewBBACAは "BSR-Based Allele Calling Algorithm "の略である。chew "の部分は "Comprehensive and Highly Efficient Workflow "とも考えられるが、現時点ではまだその主張をするには少し作業が必要なので、ソフトウェア名に "chew "を付け加えてクールさをプラスしている。BSRとは、Rasko DAらが提案したBLAST Score Ratioの略である。chewBBACAは、全ゲノムとコアゲノムのMultiLocus Sequence Typing (wg/cgMLST)スキーマの作成と検証のための一連の機能を含む包括的なパイプラインであり、マルチプロセッサの設定で実行できるBlast Score Ratioに基づく対立遺伝子呼び出しアルゴリズムと、遺伝子座の対立遺伝子変異を可視化して検証するための一連の機能を提供する。
https://github.com/B-UMMI/chewBBACA/wiki
インストール
Python dependencies:
- numpy>=1.14.0
- scipy>=0.13.3
- biopython>=1.70
- plotly>=1.12.9
- SPARQLWrapper>=1.8.0
Other dependencies:
- BLAST 2.5.0+ or above
- Prodigal 2.6.0 or above
#conda (bioconda link)
conda install -c bioconda chewbbaca
#pip
pip3 install chewbbaca
> chewBBACA.py -h
$ chewBBACA.py -h
chewBBACA version: 2.5.6
Authors: Mickael Silva, Pedro Cerqueira, Rafael Mamede
Github: https://github.com/B-UMMI/chewBBACA
Wiki: https://github.com/B-UMMI/chewBBACA/wiki
Tutorial: https://github.com/B-UMMI/chewBBACA_tutorial
Contacts: imm-bioinfo@medicina.ulisboa.pt
USAGE: chewBBACA.py [module] -h
Select one of the following functions:
CreateSchema: Create a gene-by-gene schema based on a set of input genomes.
AlleleCall: Determine the allelic profiles of a set of input genomes based on a schema.
SchemaEvaluator: Tool that builds an html output to better navigate/visualize your schema.
TestGenomeQuality: Analyze your allele call output to refine schemas.
ExtractCgMLST: Determines the set of loci that constitute the core genome based on a threshold.
RemoveGenes: Remove a provided list of loci from your allele call output.
PrepExternalSchema: Adapt an external schema to be used with chewBBACA.
JoinProfiles: Join two profiles in a single profile file.
UniprotFinder: Retrieve annotations for loci in a schema.
DownloadSchema: Download a schema from the Chewie-NS.
LoadSchema: Upload a schema to the Chewie-NS.
SyncSchema: Synchronize a schema with its remote version in the Chewie-NS.
NSStats: Retrieve basic information about the species and schemas in the Chewie-NS.
> chewBBACA.py CreateSchema -h
$ chewBBACA.py CreateSchema -h
chewBBACA version: 2.5.6
Authors: Mickael Silva, Pedro Cerqueira, Rafael Mamede
Github: https://github.com/B-UMMI/chewBBACA
Wiki: https://github.com/B-UMMI/chewBBACA/wiki
Tutorial: https://github.com/B-UMMI/chewBBACA_tutorial
Contacts: imm-bioinfo@medicina.ulisboa.pt
usage:
Create schema from input genomes:
chewBBACA.py CreateSchema -i <input_files> -o <output_directory> -ptf <ptf_path>
Create schema from input genomes with non-default parameters:
chewBBACA.py CreateSchema -i <input_files> -o <output_directory> --ptf <ptf_path>
--cpu <cpu_cores> --bsr <blast_score_ratio> --l <minimum_length>
--t <translation_table> --st <size_threshold>
Create schema from single FASTA file:
chewBBACA.py CreateSchema -i <input_file> -o <output_directory> --ptf <ptf_path> --CDS
Creates a wgMLST schema based on a set of input genomes.
positional arguments:
CreateSchema
optional arguments:
-h, --help show this help message and exit
-i [INPUT_FILES] Path to the directory that contains the input FASTA
files. Alternatively, a single file with a list of
paths to FASTA files, one per line (default: None)
-o OUTPUT_DIRECTORY Output directory where the schema will be created.
(default: None)
--ptf PTF_PATH Path to the Prodigal training file. (default:
False)
--bsr BLAST_SCORE_RATIO BLAST Score Ratio value. Sequences with alignments
with a BSR value equal to or greater than this
value will be considered as sequences from the same
gene. (default: 0.6)
--l MINIMUM_LENGTH Minimum sequence length accepted for a coding
sequence to be included in the schema. (default:
201)
--t TRANSLATION_TABLE Genetic code used to predict genes and to translate
coding sequences. (default: 11)
--st SIZE_THRESHOLD CDS size variation threshold. At the default value
of 0.2, alleles with size variation +-20 percent
will be classified as ASM/ALM. (default: 0.2)
--cpu CPU_CORES Number of CPU cores that will be used to run the
CreateSchema process (will be redefined to a lower
value if it is equal to or exceeds the totalnumber
of available CPU cores). (default: 1)
--b BLASTP_PATH Path to the BLASTp executables. (default: blastp)
--CDS Input is a FASTA file with one representative
sequence per gene in the schema. (default: False)
--v Increased output verbosity during execution.
(default: False)
> chewBBACA.py AlleleCall -h
$ chewBBACA.py AlleleCall -h
chewBBACA version: 2.5.6
Authors: Mickael Silva, Pedro Cerqueira, Rafael Mamede
Github: https://github.com/B-UMMI/chewBBACA
Wiki: https://github.com/B-UMMI/chewBBACA/wiki
Tutorial: https://github.com/B-UMMI/chewBBACA_tutorial
Contacts: imm-bioinfo@medicina.ulisboa.pt
usage:
Perform AlleleCall with schema default parameters:
chewBBACA.py AlleleCall -i <input_files> -g <schema_directory> -o <output_directory>
Perform AlleleCall with non-default parameters:
chewBBACA.py AlleleCall -i <input_files> -g <schema_directory> -o <output_directory> --ptf <ptf_path>
--cpu <cpu_cores> --bsr <blast_score_ratio> --l <minimum_length>
--t <translation_table> --st <size_threshold>
Performs allele calling to determine the allelic profiles of a set of input
genomes. The process identifies new alleles, assigns an integer identifier to
those alleles and adds them to the schema.
positional arguments:
AlleleCall
optional arguments:
-h, --help show this help message and exit
-i [INPUT_FILES] Path to the directory with the genomes FASTA files
or to a file with a list of paths to the FASTA
files, one per line. (default: None)
-g SCHEMA_DIRECTORY Path to the schema directory with the genes FASTA
files. (default: None)
-o OUTPUT_DIRECTORY Output directory where the allele calling results
will be stored. (default: None)
--ptf PTF_PATH Path to the Prodigal training file. Default is to
get training file from schema directory. (default:
False)
--bsr BLAST_SCORE_RATIO BLAST Score Ratio value. Sequences with alignments
with a BSR value equal to or greater than this
value will be considered as sequences from the same
gene. (default: 0.6)
--l MINIMUM_LENGTH Minimum sequence length accepted for a coding
sequence to be included in the schema. (default:
201)
--t TRANSLATION_TABLE Genetic code used to predict genes and to translate
coding sequences (default=11). (default: 11)
--st SIZE_THRESHOLD CDS size variation threshold. At the default value
of 0.2, alleles with size variation +-20 percent
will be classified as ASM/ALM (default: 0.2)
--cpu CPU_CORES Number of CPU cores/threads that will be used to
run the CreateSchema process (will be redefined to
a lower value if it is equal to or exceeds the
totalnumber of available CPU cores/threads).
(default: 1)
--b BLASTP_PATH Path to the BLASTp executables. (default: blastp)
--json Output report in JSON format. (default: False)
--fc Continue allele call process that was interrupted.
(default: False)
--fr Force process reset even if there are temporary
files from a previous process that was interrupted.
(default: False)
--db If the profiles in the output matrix should be
stored in the local SQLite database. (default:
True)
--v Increased output verbosity during execution.
(default: False)
It is strongly advised to perform AlleleCall with the default schema
parameters to ensure more consistent results.
> chewBBACA.py RemoveGenes -h
$ chewBBACA.py RemoveGenes -h
chewBBACA version: 2.5.6
Authors: Mickael Silva, Pedro Cerqueira, Rafael Mamede
Github: https://github.com/B-UMMI/chewBBACA
Wiki: https://github.com/B-UMMI/chewBBACA/wiki
Tutorial: https://github.com/B-UMMI/chewBBACA_tutorial
Contacts: imm-bioinfo@medicina.ulisboa.pt
usage:
Remove a set of genes from a matrix with allelic profiles:
chewBBACA.py RemoveGenes -i <input_file> -g <genes_list> -o <output_file>
Remove loci from a matrix with allelic profiles.
positional arguments:
RemoveGenes Remove loci from a matrix with allelic profiles.
optional arguments:
-h, --help show this help message and exit
-i INPUT_FILE TSV file that contains a matrix with allelic profiles
determined by the AlleleCall process.
-g GENES_LIST File with the list of genes to remove.
-o OUTPUT_FILE Path to the output file that will be created with the new
matrix.
--inverse List of genes that is provided is the list of genes to keep
and all other genes should be removed.
> chewBBACA.py TestGenomeQuality -h
$ chewBBACA.py TestGenomeQuality -h
chewBBACA version: 2.5.6
Authors: Mickael Silva, Pedro Cerqueira, Rafael Mamede
Github: https://github.com/B-UMMI/chewBBACA
Wiki: https://github.com/B-UMMI/chewBBACA/wiki
Tutorial: https://github.com/B-UMMI/chewBBACA_tutorial
Contacts: imm-bioinfo@medicina.ulisboa.pt
usage:
Evaluate genome quality with default parameters:
chewBBACA.py TestGenomeQuality -i <input_file> -n <max_iteration> -t <max_threshold>-s <step>
This process evaluates the quality of genomes based on the results of the
AlleleCall process.
positional arguments:
TestGenomeQuality Evaluate the quality of input genomes based on allele
calling results.
optional arguments:
-h, --help show this help message and exit
-i INPUT_FILE Path to file with a matrix of allelic profiles.
(default: None)
-n MAX_ITERATION Maximum number of iterations. (default: None)
-t MAX_THRESHOLD Maximum threshold of bad calls above 95 percent.
(default: None)
-s STEP Step between each threshold analysis. (default: None)
-o OUTPUT_DIRECTORY Path to the output directory that will store output
files (default: .)
-v, --verbose Increase stdout verbosity. (default: False)
> chewBBACA.py ExtractCgMLST -h
$ chewBBACA.py ExtractCgMLST -h
chewBBACA version: 2.5.6
Authors: Mickael Silva, Pedro Cerqueira, Rafael Mamede
Github: https://github.com/B-UMMI/chewBBACA
Wiki: https://github.com/B-UMMI/chewBBACA/wiki
Tutorial: https://github.com/B-UMMI/chewBBACA_tutorial
Contacts: imm-bioinfo@medicina.ulisboa.pt
usage:
Determine cgMLST:
chewBBACA.py ExtractCgMLST -i <input_file> -o <output_directory>
Determine cgMLST based on non-default threshold:
chewBBACA.py ExtractCgMLST -i <input_file> -o <output_directory>
--p <threshold>
Remove genes and genomes from matrix:
chewBBACA.py ExtractCgMLST -i <input_file> -o <output_directory>
--r <genes2remove> --g <genomes2remove>
Determines the set of loci that constitute the core genome based on a
threshold.
positional arguments:
ExtractCgMLST Determines the set of loci that
constitute the core genome based on a
threshold.
optional arguments:
-h, --help show this help message and exit
-i INPUT_FILE Path to input file containing a
matrix with allelic profiles.
(default: None)
-o OUTPUT_DIRECTORY Path to the directory where the
process will store output files.
(default: None)
--p THRESHOLD, -p THRESHOLD Genes that constitute the core genome
must be in a proportion of genomes
that is at least equal to this value.
(default: 1)
--r GENES2REMOVE, -r GENES2REMOVE Path to file with a list of
genes/columns to remove from the
matrix (one gene identifier per
line). (default: False)
--g GENOMES2REMOVE, -g GENOMES2REMOVE Path to file with a list of
genomes/rows to remove from the
matrix (one genome identifier per
line). (default: False)
実行方法
スキーマとは、MLST解析で使用される、あらかじめ定義された遺伝子座のセットのことである。従来のMLSTのスキーマは、ハウスキーピング遺伝子の内部断片である7つの遺伝子座に依存しており、各遺伝子座はプライマーを用いて増幅され、定義されたサイズの断片が得られることで定義されていた。ゲノム解析では、スキーマとは、遺伝子座の集合のことである。
スキーマの作成には、通常、95%の株に存在するというしきい値が用いられる。これは、配列のカバレッジの問題、アセンブリの問題、またはドラフトゲノムアセンブリの使用に関連するその他の問題のために、5%までのアレルが同定されない可能性があるためである。
1、prodigalのトレーニングファイルを作成する。ハイクオリティなゲノムfastaを使用することが推奨されている。
prodigal -i input-genome.fasta -t training_file.trn -p single
training_file.trnが出力される。
2、スキーマの作成
genomeのFASTAディレクトリと、1で得たprodigalのトレーニングファイルを指定する。
chewBBACA.py CreateSchema -i genome_fasta_dir/ -o schema_dir --cpu 12 --ptf training_file.trn
all versus all BLASTP検索を実行し、Blast Score Ration(BSR)を計算することにより、すべてのCDSを一意の座位にグループ化する。BSRのペアワイズ比較が0.6より大きいCDSは、同じ遺伝子座の対立遺伝子とみなされ、大きい方の対立遺伝子(bp単位)がリストに保持される。 wgMLSTスキーマが出力される。
3、アレルコール
chewBBACA.py AlleleCall -i genome_fasta_dir/ -g schema_dir/ -o outdir/ --cpu 12 --ptf training_file.trn
- --bsr BLAST_SCORE_RATIO BLAST Score Ratio value. Sequences with alignments with a BSR value equal to or greater than this value will be considered as sequences from the same gene. (default: 0.6)
デフォルトではBSR閾値0.6が使用される。

出力

results_alleles.tsv

4、パラログ検出とフィルタリング
chewBBACA.py RemoveGenes -i outdir/results_20201129T173501/results_alleles.tsv -g outdir/results_20201129T173501/RepeatedLoci.txt -o alleleCallMatrix_cg
===========================
chewBBACA - RemoveGenes
===========================
Provided list has 35 genes.
Removing 35 genes...done.
アレルコールファイルalleleCallMatrix_cg.tsvが出力される。
5、評価する。
chewBBACA.py TestGenomeQuality -i alleleCallMatrix_cg.tsv -n 13 -t 200 -s 5

5、wgMLSTのアレルコール後、cgMLSTコールも行うために95%でフィルタリング
chewBBACA.py ExtractCgMLST -i alleleCallMatrix_cg.tsv -o cgMLST_completegenomes -p 0.95
cgMLSTは95%から99%のゲノムで存在すると定義されているため、wgMLSTより数は少なくなる(補助的な遺伝子座を削除する)。wgMLSTのアレルコール後にこの閾値でフィルタリングしてcgMLSTのコールにする。
=============================
chewBBACA - ExtractCgMLST
=============================
Building presence and absence matrix...done.
Determining genes in the core genome...done.
Determining missing data per genome...done.
Core genome composed of 1264/3095 genes.
6、cgMLST_completegenomes/cgMLST.tsv を wgMLST出力のresults_all/results_〜/results_alleles.tsv のアレルコール結果と連結する。
chewBBACA.py JoinProfiles -p1 cgMLST_completegenomes/cgMLST.tsv -p2 outputDirectory/results_20201129T173501/results_alleles.tsv -o cgMLST_all.tsv
============================
chewBBACA - JoinProfiles
============================
reading profile 1
reading profile 2
building new profile
Done
cgMLST_all.tsvが出力される。
7、cgMLSTの結果を評価する。
chewBBACA.py TestGenomeQuality -i alleleCallMatrix_cg.tsv -n 13 -t 200 -s 5
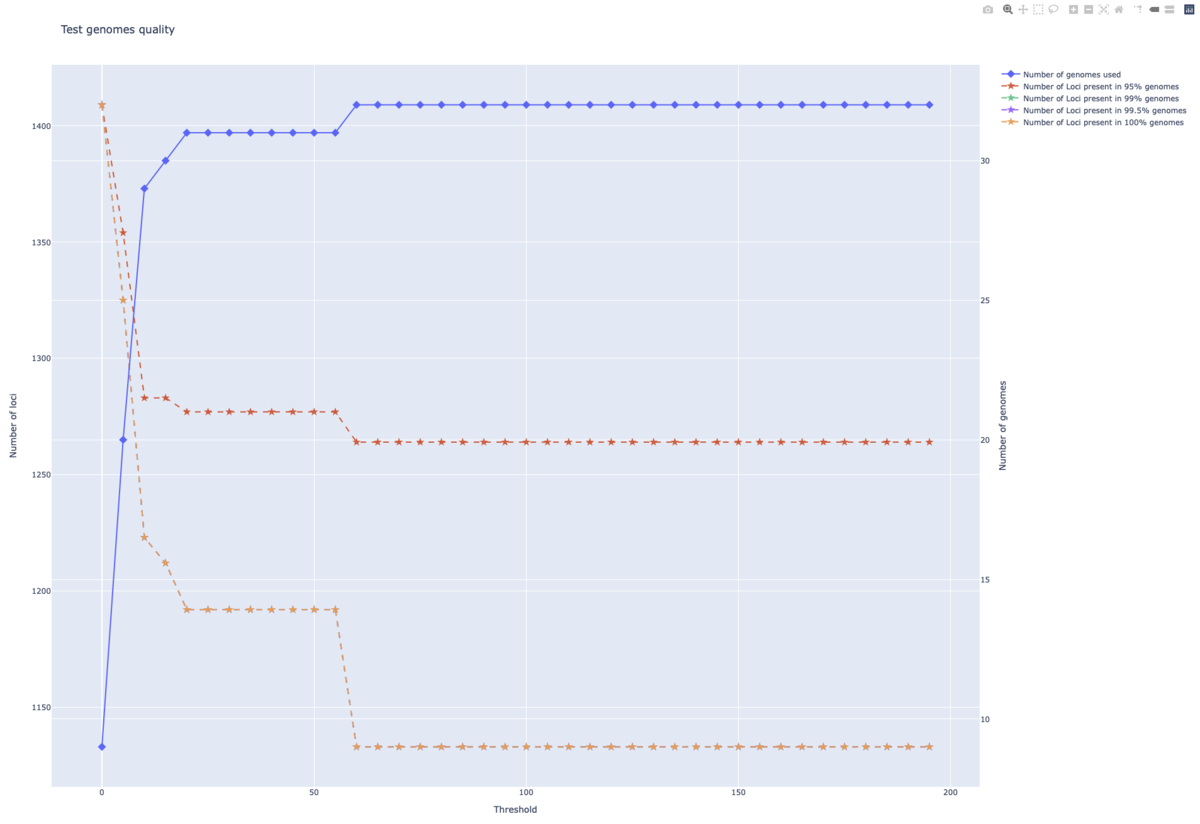
8、スキーマの評価
chewBBACA.py ExtractCgMLST -i cgMLST_all.tsv -o cgMLST_25 -g removedGenomes.txt
=============================
chewBBACA - ExtractCgMLST
=============================
Building presence and absence matrix...done.
Determining genes in the core genome...done.
Determining missing data per genome...done.
Core genome composed of 1133/1264 genes.
出力

Presence_Absence.tsv

PHYLOViZ OnlineでMinimum Spanning Tree として視覚化する
https://online.phyloviz.net/index

cgMLST_all.tsvを読み込ませる。ログイン不要。
距離行列を出力 =>
すでにトレーニングファイルが用意されている種もあります。GitHub - B-UMMI/chewBBACA: BSR-Based Allele Calling AlgorithmのQ&Aを確認して下さい。
テストラン
(link) Streptococcus agalactiaeの32の完全長ゲノムと 682のSRA由来アセンブリ配列のMLST解析。以下では32ゲノムのwgMLSTの例のみ記載。
git clone https://github.com/B-UMMI/chewBBACA_tutorial
cd chewBBACA_tutorial/
unzip genomes/complete_genomes.zip
#1 wgMLSTスキーマの作成(chromosomeのみ含まれる)
chewBBACA.py CreateSchema -i complete_genomes/ --cpu 6 -o schema_seed --ptf Streptococcus_agalactiae.trn
#2 Allele calling
chewBBACA.py AlleleCall -i complete_genomes/ -g schema_seed/ -o results_cg --cpu 6 --ptf Streptococcus_agalactiae.trn
#3 Paralog detection
chewBBACA.py RemoveGenes -i results_cg/results_20201129T181346/results_alleles.tsv -g results_cg/results_20201129T181346/RepeatedLoci.txt -o alleleCallMatrix_cg
他に682個の Streptococcus agalactiae assemblies を使ってcgMLSTを調べるチュートリアルがあります。
引用
chewBBACA: A complete suite for gene-by-gene schema creation and strain identification Open Access
Mickael Silva1, Miguel P. Machado, Diogo N. Silva, Mirko Rossi, Jacob Moran-Gilad, Sergio Santos1, Mario Ramirez, João André Carriço
Microb Genom. 2018 Mar;4(3)
関連
闘病していた親友が亡くなってしまったので数日お休みをもらいました。まだドタバタしていますが、心の整理がついてきたので今日から再開します。よろしくお願いします。
健康はとっても大事です。皆様、何事も無理はしすぎず自分のできる範囲で行って下さい。