2022/03/28 URL 更新, POCP matrix追記
Bergey's Manual of Systematics of Archaea and Bacteriaより
次世代シークエンシングアプローチの展開により、完全にシークエンシングされたゲノムの数は急速に増加している。その結果、単一ゲノムだけでなく、関連する大規模なゲノム群を比較的に解析することが可能になった。また、タイプストレインのゲノムの全ゲノムシークエンシングは、より高分解能の系統分類・分類学的分類を得るための大きな可能性を秘めている。EDGARプラットフォームは、過去9年間で比較ゲノムの分野で最も確立されたソフトウェアツールの一つとなった。この間、ソフトウェアは継続的に改良され、多くの新しい解析機能が追加されてきた。近年では、コアゲノムに基づく系統図・分類学的解析にEDGARを利用することが、このソフトウェアの主な応用分野となっている。原核生物種のすべてのタイプストレインのゲノム配列を生成することに焦点を当てることで、基本的な16S rRNA遺伝子配列の系統図を大幅に拡張し、より高分解能のコアゲノムベースの分類学に拡張することができ、ラボでの集中的なDNA-DNAハイブリダイゼーション(DDH)を、DDHと同様に種の境界を反映するゲノム配列ベースの指標に置き換えることができる。
EDGARのウェブベースのユーザーインターフェースは、新種の提案に必要な系統学的な種間・種内分類学的解析に必要なすべてのツールを提供している。EDGARは、隣接結合法と最尤法を用いてコアゲノムベースの系統樹を計算し、アミノ酸同一性(AI)と平均ヌクレオチド同一性(ANI)マトリックスを計算する。さらに、ベン図、シンテニープロット、直交遺伝子のゲノム近傍の比較表示などの便利な可視化機能を提供する。また、統計解析、レプリコングループ化オプション、メタ遺伝子セットの第2レベル解析など、様々な新機能が追加された。進化の関係を迅速に調査することができ、近親ゲノムの差異遺伝子の内容についての新たな生物学的洞察を得るプロセスを簡略化することができる。
また、EDGARでは、比較ゲノムや系統図の結果を事前に計算したプロジェクトを公開データベースとして提供している。このプラットフォームでは、8,079の完全ゲノムからなる322の属ベースの公開データベースを提供している。ここでは、これらの属ベースのプロジェクトの他に、ファミリーレベルでクラスタリングされた226の新しい公開プロジェクトを紹介する。これらの新規公開プロジェクトは、さらに4,400のゲノムを含んでいる。EDGARは学術利用は無料で、ドイツのバイオインフォマティクス・インフラストラクチャ・ネットワーク(de.NBI)のサービスとして提供されている。EDGARは公開ウェブサーバー http://edgar.computational.bio から利用できる。
EAGAR2の論文より
微生物のゲノム配列の利用可能性が急速に高まっていることから、密接に関連したゲノムの比較に基づく機能解析を支援するバイオインフォマティクスソフトウェアツールの需要が高まっている。遺伝子レベルでの比較アプローチを利用することで、研究対象となる生物の共通の特徴を表すコア遺伝子の洞察を得ることができる。また、逆にシングルトン遺伝子を同定することで、個々のゲノムの特異的な性質を明らかにすることができる。EDGARプラットフォームは、最初の出版以来、比較ゲノムの分野で最も確立されたソフトウェアツールの一つとなっている。ここ数年、EDGARは継続的に改良され、多くの新しい解析機能が追加されてきた。新バージョンのEDGAR 2.0では、遺伝子オルソロジー推定アプローチが新たに設計され、完全に再実装された。他の新機能の中でも、EDGAR 2.0では、AAI(Average Amino Acid Identity)やANI(Average Nucleotide Identity)行列のような系統解析機能の拡張、ゲノムセットサイズの統計、インタラクティブなシンテニープロットやベン図のような最新の可視化機能を提供している。これにより、微生物ゲノム間の進化関係を迅速かつユーザーフレンドリーに調査することが可能となり、微生物ゲノムに含まれる遺伝子の違いについての新たな生物学的洞察を得るプロセスを簡素化することができる。すべての機能は、ウェブベースのプラットフォームに依存しないユーザーインターフェースを介して研究者に提供されており、事前に計算されたデータセットを簡単に閲覧することができる。ウェブサーバーは http://edgar.computational.bio からアクセスできる。
help
https://www.uni-giessen.de/fbz/fb08/Inst/bioinformatik/software/EDGAR/Features
https://edgar3.computational.bio.uni-giessen.de/cgi-bin/edgar_login.cgiにアクセスする。

ここではサルモネラを選択。

まずはコアゲノムを探索してみる。パンゲノムも同じ感じに使えるが、コアゲノムとは異なり、パンゲノム解析では、特定のゲノムにしか見つからない遺伝子もプリントされる。
左のメニューからコアゲノムを選択。

SELECT ALLを選択するかクリックしてゲノムを選ぶALLの表記はchrとplasmid全てを含む。参照のゲノムは一度選択後に右クリックすることで選べる。

CP015724を選んだ。リファレンスのゲノムはより黒い色に変わる。
SHOW CORE GENOMEボタンを押すと解析がスタートする。 ゲノムが多すぎると計算が終わらないことがあるので注意。
出力は遺伝子のリストとなる。1行に1セットのオーソロガス遺伝子が表示される。参照のゲノムは結果は1列目に表示される。パンゲノム解析では、特定の遺伝子のオルソログが他のゲノムに見当たらない場合はセルに"-"が表示される。

各行の最後の2列(下図)には、その行の遺伝子のマルチプルアラインメントと、翻訳開始部位の前の400bpの上流領域のマルチプルアラインメントを作成するためのリンクになっている。DNAまたはアミノ酸配列どちらでも作成できる。

遺伝子はFASTAファイル(DNAまたはタンパク質配列)や座 標タグと機能記述をTABで区切ったフラットファイルとして保存できる。

他の解析も、同じようにゲノムを選んでスタートさせる。

結果だけ順番に見ていく。
Upset plot

Venn diagram

GET LISTから結果のリストをダウンロードできる。
ゲノムの数が多すぎると視覚化結果はよくわからなくなるので注意。その場合は複雑な部分集合を調べることができるCalculate genesetsを使う。
Circular Plot
上では3ゲノム選択して描画した。


Core development plot
より多い数のゲノムに対して「本当の」コアゲノムのサイズやシングルトン数を推定する。
ある特定のゲノムセットについて計算されたコアゲノムは、常にそのゲノムセットの状況のスナップショットに過ぎない。種の "本当の "コアゲノムを知るためには、無限のサイズのゲノムセットについてコア遺伝子の数を外挿する。これは、Tettelinら(2005)による近似的なアプローチによって行われる。

Singleton development plot

Pan development plot

Pan vs. Core development plot

Synteny Plots
下図のように、3つ位以上のゲノムでも実行可能。

Synteny Matrix

Create AAI matrix


Create ANI matrix

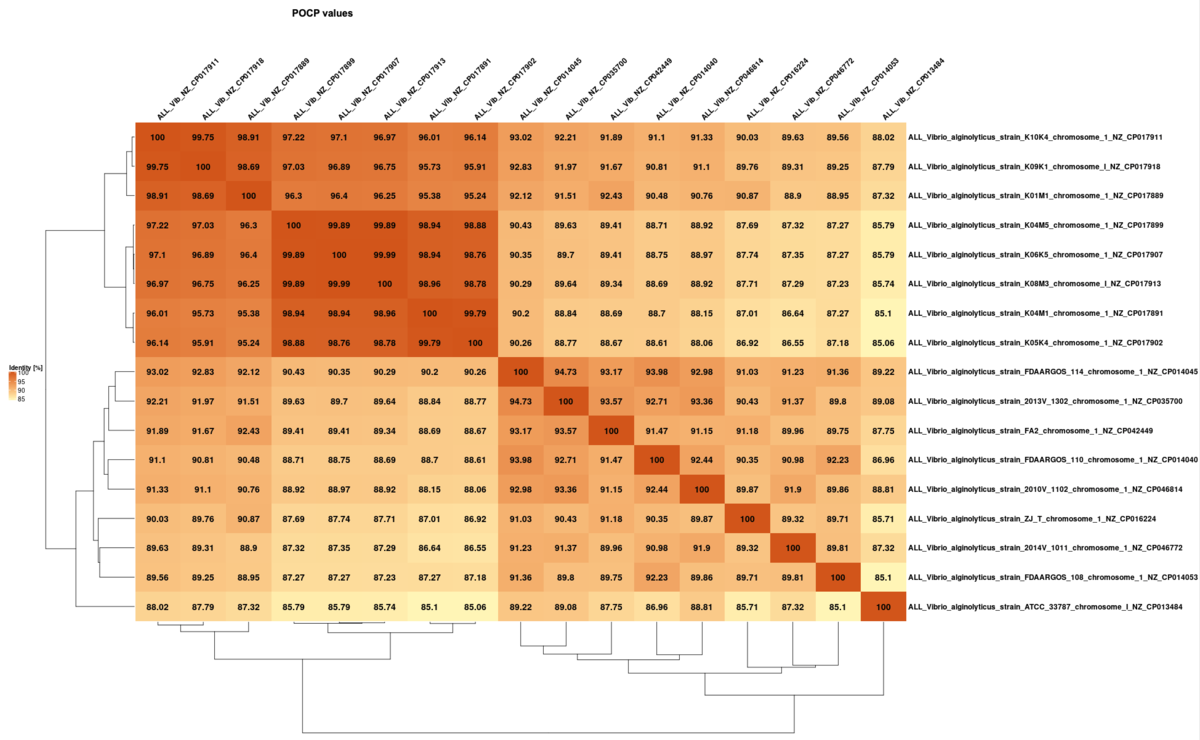
POCP (percentage of conserved genes) matrix
Phylogenetic trees
EDGARの主な目的は複数のゲノム間のオルソログの推定であるため、そのオルソログ情報から系統知識を推論することは容易にできる。このパイプラインでは、選択されたゲノムのコアゲノムを計算する。全ゲノム中に見出されたオロソロガスな遺伝子のセットは、マルチプルアラインメントツールMUSCLE(Edgar, 2004)を用いて個別にアラインメントされている。アライメントは、数十万残基になる巨大なマルチプルアラインメントに連結される。このアラインメントから距離行列を計算し、最終的にこの距離行列に基づいてNeighbor-Joining法を用いて系統樹が構築されている。後者の二つの方法は、Felsenstein (1995) による PHYLIP の実装で使用されている。Neighbor-Joining法が選択されているのは、非常に計算効率の良いヒューリスティックなアプローチであるため、コアゲノムに基づく系統樹構築の結果として得られる大規模なデータセットに適しているからである。

補足
マニュアルより
EDGARは、より高いレベルの比較のために "メタコンティグ "やレプリコングループを定義する。基本的にEDGARは単一のコンティグや完全なゲノムの比較を提供しているが、より複雑な比較が必要な場合(例えばある生物のすべてのプラスミドと別の生物のすべてのプラスミドの遺伝子内容を比較したいなど)、生物とコンティグの間の抽象化レベルが必要となる。このような場合、ユーザーが選択したコンティグのグループを作成して使用することができる。
引用
EDGAR: A Versatile Tool for Phylogenomics
Jochen Blom Stefanie P. Glaeser Tobias Juhre Julian Kreis Patrick H.G. Hanel Jascha G. Schrader Peter Kämpfer Alexander Goesmann
Bergey's Manual of Systematics of Archaea and Bacteria, First published: 06 June 2019
https://doi.org/10.1002/9781118960608.bm00038
EDGAR 2.0: an enhanced software platform for comparative gene content analyses
Jochen Blom, Julian Kreis, Sebastian Spänig, Tobias Juhre, Claire Bertelli, Corinna Ernst, Alexander Goesmann
Nucleic Acids Res. 2016 Jul 8; 44(Web Server issue): W22–W28
EDGAR: a software framework for the comparative analysis of prokaryotic genomes
Jochen Blom, Stefan P Albaum, Daniel Doppmeier, Alfred Pühler, Frank-Jörg Vorhölter, Martha Zakrzewski, Alexander Goesmann
BMC Bioinformatics. 2009 May 20;10:154
EDGAR3.0