2019 10/28 誤字修正、コメント追加、11/5 誤字修正、捕捉追加、11/6 追記
2020 2/21 インストールコマンド修正、3/4 ツイート追加、4/21 インストールの説明を修正、8/23 補足
、9/9 KBase補足、論文リンク追加、12/28 データベースダウンロードリンク更新
2021 5/9 データベースダウンロードリンク更新、9/28 論文引用
2022/3/13 CPU数変更、構成変更
Preprintより
過去数年のシーケンスされた微生物ゲノムの急速な拡大により、ゲノム配列に基づいた詳細な分類の構築が可能になった(Segata et al、2013; Hugenholtz et al、2016; Garrity et al、2016; Yoon et al、2017)。Microbial taxonomyでは、微生物の分類は、微生物の多様性を正確に記述し、科学的結果を伝えるための共通言語を提供するために不可欠である(Godfray 2002)。ゲノム配列ベースの系統樹は、進化の関係と異なる進化速度を考慮に入れた分類法を定義するための自然なフレームワークを提供する。多くの分類群が多系統のグループ分けを制限しているため、現在の微生物分類学は進化関係と矛盾することが多い。(McDonald et al., 2012)。これは、形態学的類似性を共有する微生物がクロストリジウム属として誤って分類されているclostridia(wiki)の例に示されるように、歴史的な表現型ベースの分類に一部起因している(Yutin&Galparin、2013; Beiko 2015)。現代の微生物分類学は主に16S rRNA関係によって導かれ、そのような矛盾は16S rRNA遺伝子ツリーで観察できる(McDonald et al、2012; Yilmaz et al。 2014)、しかし、タスクの規模と微生物の正式な再分類の長いプロセスのため、ほとんどは修正されていない(Yarza et al、2014)。
既存の配列スベースの微生物分類法で、2番目の明らかでない問題は、ツリー全体での階級ランクの不均一性である。集中的な研究の対象である領域は、phylogenetic depthが同等の他の部分よりも多くの分類群に分割される傾向がある。たとえば、腸内細菌科(数十の属を含む)は、ツリーの他の部分の単一の属、例えばBacillusと同じしかない。逆に、研究されていないグループはしばしばひとまとめにされる。たとえば、現在Synergistetes門は単一のファミリーによって表されているが(Jumas-Bilak et al、2009)、50以上の集中的に研究されたツリーの部分では複数ファミリーレベルのグループを構成している。Yarza et al. (2014) による最近の研究では、16S rRNA配列同一性しきい値を使用して分類ランクを標準化することを提案し、これらのしきい値と既存の分類法との間の高い割合の不一致を報告している。
16S rRNA遺伝子の関係に基づく現在の微生物分類法には様々な限界があり、これには、highestの階級とlowestの階級での低い phylogenetic resolution(Case et al、2007; Janda et al、2007)、プライマーのミスマッチによる多様性の欠如(Schulz et al、2017)、およびPCRが生成するキメラ配列によって異種のグループがまとめられ、ツリートポロジーが破損する可能性(DeSantis et al、2006)、が含まれる。(翻訳が怪しいため複数文章省略)
ここでは、未培養生物(メタゲノム合成または単一細胞ゲノム)13,636(14.4%)を含む94,759の細菌ゲノムをカバーする120 ubiquitous single-copy proteinsのconcatenationから推論された、系統学に基づいたbacterial taxonomyを提案する。この分類内のTaxonomic groupsは、系統固有の進化率を正規化した後、類似のphylogenetic depths のmonophyleticな系統を記述する。我々(著者ら)はこれをGTDB taxonomyと呼び、Genome Taxonomyデータベースウェブサイト(http://gtdb.ecogenomic.org)で公開している。
2023/03/23
Bergey's Manualへのリンクが追加されました。
GTDB is on a mission to promote excellence in research by linking its content with the iconic taxonomic resource - Bergey's Manual of Systematics of Archaea and Bacteria.
— GTDB (@ace_gtdb) March 22, 2023
Access to the chapters is available in our Taxonomy Tree viewer via your institutional subscription. pic.twitter.com/T56OlSOU4d
アーキアが追加されました。
Preprint is out - "A rank-normalized archaeal taxonomy based on genome phylogeny resolves widespread incomplete and uneven classifications" - based on #GTDB @ace_gtdb with @m_chuvochina @PhilHugenholtz @donovan_parks @d_w_waite B. Whitman and others https://t.co/3meuYpHRLs
— Chris Rinke (@ChristianRinke) 2020年3月4日
GTDB-Tk v0.3.2 has been released. It features improved output tables and a ~40 min reduction in classification time. This version is the basis of the upcoming GTDB-Tk manuscript and was validated on >10,000 MAGs. https://t.co/mCZrEgXMdr
— GTDB (@ace_gtdb) July 12, 2019
You can now visualize the history of GTDB taxa: https://t.co/oqXCJjLifG. History of Bacillus pumilus illustrates the impact of the ANI species clustering introduced in R04-RS89, including classification of 16 genomes lacking species assignments at NCBI. pic.twitter.com/Vjazjes5kY
— GTDB (@ace_gtdb) October 23, 2019
Preprint describing the methodology used to establish species clusters in the GTDB is out in @biorxivpreprint: https://t.co/G6QYarZ5ue pic.twitter.com/VGRHsiYmU1
— GTDB (@ace_gtdb) September 19, 2019
GTDB-Tkの使い方
GTDB-Tkは、客観的にtaxonomic classificationsをバクテリアおよびアーキアのゲノムにアサインするためのソフトウェアツールキットである。 これは、最近の進歩で出現してきた環境サンプルから直接取得し、シーケンシングとアセンブリによって得た数百または数千のメタゲノムアセンブリゲノム(MAG)で機能するよう設計されている。 単離株およびシングルセルゲノムにも適用できる。 GTDB-Tkはオープンソースで、GNU General Public License(バージョン3)の下でリリースされている。
現在、GTDB-Tkは活発に開発および検証中となっている。 使用する場合、ツリーを個別に手動で調べ、GTDB-Tkの予測に不一致があれば注意する事。GTDB-Tkのリリースに関する通知は、GTDBのTwitterアカウント(https://twitter.com/ace_gtdb)から入手できる。
GTDB-Tk FAQ
https://github.com/Ecogenomics/GTDBTk/blob/stable/docs/faq.md
インストール
ubuntu18.0.4のpython3環境でテストした(メモリ512GBのサーバー使用)。
依存
Hardware requirements
- ~100Gb of memory to run
- ~27Gb of storage
- ~1 hour per 1,000 genomes when using 64 CPUs
Python libraries
GTDB-Tk is designed for Python 2.7 and requires the following Python libraries:
- dendropy >=4.1.0: Python library for phylogenetics.
- SciPy Stack: at least the Matplotlib, NumPy, and SciPy libraries.
Third-party software
GTDB-Tk makes use of the following 3rd party dependencies and assumes these are on your system path:
- Prodigal >= 2.6.2: Hyatt D, et al. 2012. Gene and translation initiation site prediction in metagenomic sequences. Bioinformatics, 28, 2223-2230.
- HMMER >= 3.1: Eddy SR. 2011. Accelerated profile HMM searches. PLoS Comp. Biol., 7, e1002195.
- pplacer >= 1.1: Matsen F, et al. 2010. pplacer: linear time maximum-likelihood and Bayesian phylogenetic placement of sequences onto a fixed reference tree. BMC Bioinformatics, 11, 538.
- FastANI >= 1.0: Jain C, et al. 2017. High-throughput ANI Analysis of 90K Prokaryotic Genomes Reveals Clear Species Boundaries.bioRxiv. 256800.
- FastTree >= 2.1.9: Price MN, et al. 2010 FastTree 2 -- Approximately Maximum-Likelihood Trees for Large Alignments. PLoS ONE, 5, e9490.
本体 Github
#またはbiocondaで導入(link)
#ここでは2.7の仮想環境に依存も含めて導入
#2019 12/6 コードがpython3になった。python3環境に導入可能。
mamba create -n GTDB-Tk -c bioconda -y hmmer prodigal pplacer fastani fasttree gtdbtk
conda activate GTDB-Tk
#2021 5/9 gtdbtk v1.5.0のみ導入
mamba install -c bioconda gtdbtk=1.5.0
#pipの場合
pip install gtdbtk
dockerイメージも用意されている(データベースは別にダウンロードする必要がある)。
docker pull aaronmussig/gtdbtk
docker run --rm -itv $PWD:/data/ --rm aaronmussig/gtdbtk
> gtdbtk -h
$ gtdbtk -h
...::: GTDB-Tk v0.3.2 :::...
Workflows:
classify_wf -> Classify genomes by placement in GTDB reference tree
(identify -> align -> classify)
de_novo_wf -> Infer de novo tree and decorate with GTDB taxonomy
(identify -> align -> infer -> root -> decorate) [In Development]
Methods:
identify -> Identify marker genes in genome
align -> Create multiple sequence alignment
infer -> Infer tree from multiple sequence alignment
classify -> Determine taxonomic classification of genomes
root -> Root tree using an outgroup
decorate -> Decorate tree with GTDB taxonomy [In Development]
Tools:
trim_msa -> Trim an untrimmed MSA file based on a mask
export_msa -> Export the untrimmed archaeal or bacterial MSA file
Testing:
test -> Test the classify_wf pipeline with 3 archaeal genomes
check_install -> Verify if all GTDB-Tk data files are present
Use: gtdbtk <command> -h for command specific help
> gtdbtk classify_wf -h
$ gtdbtk classify_wf -h
usage: gtdbtk classify_wf (--genome_dir GENOME_DIR | --batchfile BATCHFILE)
--out_dir OUT_DIR [-x EXTENSION]
[--min_perc_aa MIN_PERC_AA] [--prefix PREFIX]
[--cpus CPUS] [--force] [--scratch_dir SCRATCH_DIR]
[-r] [-d] [-h]
mutually exclusive required arguments:
--genome_dir GENOME_DIR
directory containing genome files in FASTA format
--batchfile BATCHFILE
file describing genomes - tab separated in 2 columns
(FASTA file, genome ID)
required named arguments:
--out_dir OUT_DIR directory to output files
optional arguments:
-x, --extension EXTENSION
extension of files to process, gz = gzipped (default:
fna)
filter genomes with an insufficient percentage of AA
in the MSA (default: 10)
--prefix PREFIX desired prefix for output files (default: gtdbtk)
--cpus CPUS number of CPUs to use (default: 1)
--force continue processing if an error occurrs on a single
genome
--scratch_dir SCRATCH_DIR
Reduce memory usage by writing to disk (slower).
-r, --recalculate_red
recalculate RED values based on the reference tree and
all added user genomes
-d, --debug create intermediate files for debugging purposes
-h, --help show help message
データベースの準備
27GBほどのサイズがある。#テスト時はrelease89だった。=> 95 にアップデート => 202にアップデート
wget https://data.ace.uq.edu.au/public/gtdb/data/releases/release95/95.0/auxillary_files/gtdbtk_r95_data.tar.gz
tar -xvzf gtdbtk_r95_data.tar.gz
#PATHの設定
export GTDBTK_DATA_PATH=/PATH/to/release95/
#2021 5/9 r202 (GTDBtk v1.5.0が必要)
wget https://data.ace.uq.edu.au/public/gtdb/data/releases/release202/202.0/genomic_files_reps/gtdb_genomes_reps_r202.tar.gz
tar -xvzf gtdb_genomes_reps_r202.tar.gz
export GTDBTK_DATA_PATH=/PATH/to/release202/
#2022/04/12
wget https://data.gtdb.ecogenomic.org/releases/release207/207.0/auxillary_files/gtdbtk_r207_data.tar.gz
tar -xvzf gtdbtk_r207_data.tar.gz
=> release207ができる
#ダウンロードスクリプト(link)
download-db.sh
インストールチェック
gtdbtk check_install
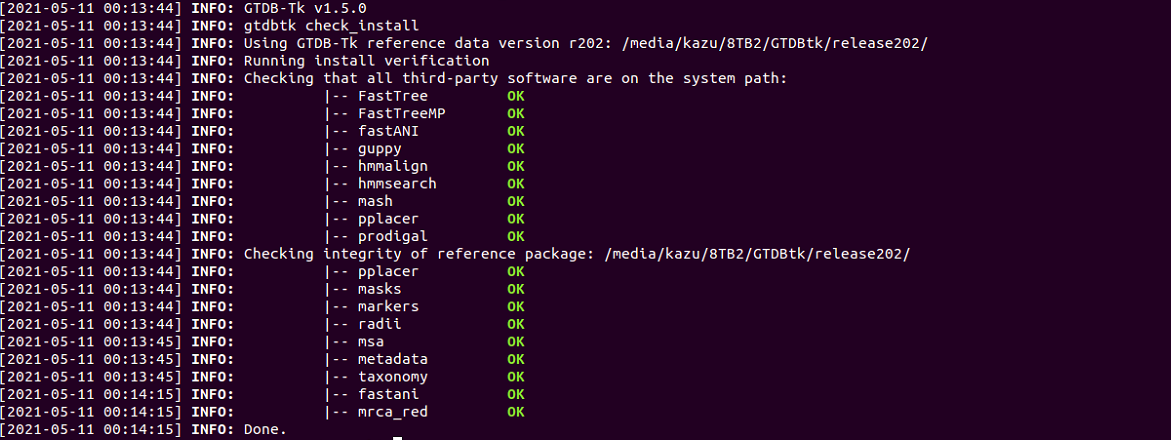
r202 (GTDBtk v1.5使用)
テストラン
gtdbtk test --out_dir OUT_DIR --cpus 10

O.K !
実行方法
1、classify_wf Classify genomes by placement in GTDB reference tree (identify -> align -> classify)
classifyワークフローは、同定、アラインメント、分類の3つのステップで構成されている。同定ステップではProdigalを使用して遺伝子コールし、HMMERパッケージを使用して系統学的推論に使用される120の細菌および122の古細菌マーカー遺伝子を同定する。アラインメントステップでは、 マーカー遺伝子をそれらのそれぞれのHMMモデルにアラインすることによってマルチプルシーケンスアラインメント(MSA)を得る。 アラインメントされたマーカー遺伝子を連結しそして連結されたMSAを約5,000アミノ酸にフィルタリングする。 最後に、分類ステップはpplacerを使用してGTDB-Tkリファレンスツリー内の各ゲノムの最尤配置を見つける。HTDB-Tkは、リファレンスゲノムにおけるその配置、その相対的進化的発散、および/またはリファレンスゲノムに対する平均ヌクレオチド同一性(ANI)に基づいて各ゲノムを分類する。
分類する(ドラフト)ゲノムのFASTA(gzip with the extension .gz is acceptable)を含むディレクトリを指定する(*2)。binned-fasta群のように複数fastaを含む場合もディレクトリを指定すればいい。
gtdbtk classify_wf --genome_dir genome_bir/ --out_dir output \
--cpus 12 --extension fasta
- --prefix desired prefix for output files (default: gtdbtk)
- --cpus number of CPUs to use (default: 1)
- --min_perc_aa filter genomes with an insufficient percentage of AA in the MSA (default: 10)
注意
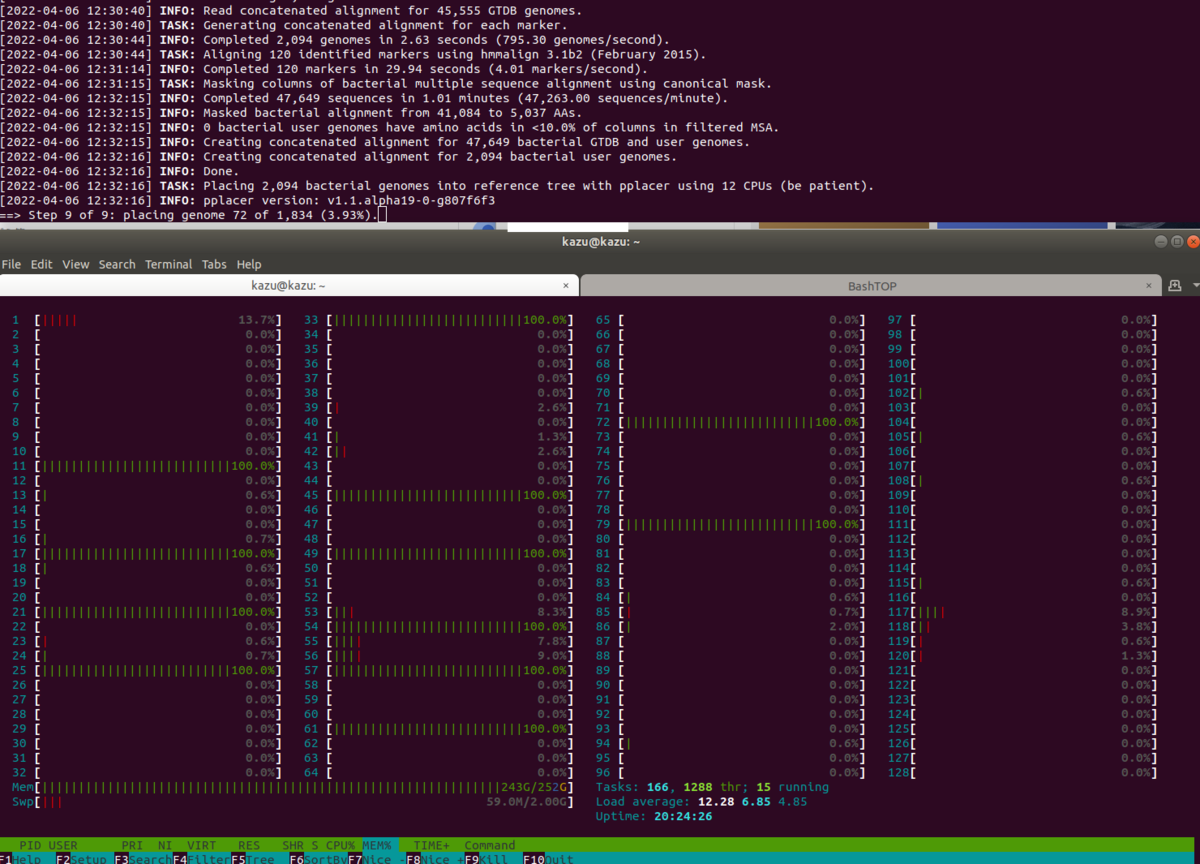
データベースが大きくなったためかもしれませんが、最新のデータベースを使うと、メモリ256GB利用できる計算機でも --cpus 60 の設定ではメモリが足らずエラーになりました。--cpus 20だとランできました。
step9が特にギリギリです。release202ではstep9でメモリを243GBほど使っています。次のバージョンアップがあれば足りなくなると予想されます。
出力

最終のclassify出力ディレクトリ。pplacerによって実行された、指定したゲノムとGTDBのreference treeの Newick formatの分子系統樹、 サマリーファイルなどが含まれる。

出力についてはGithubを参照してください。全出力について解説があります。
FastANIによりANI比較して、GTDBリファレンスツリーの既存の種へ分類します。既存の種へ分類されるには、ANI value 95%以上(*1)、アラインメントカバー率65%以上の両方を満たす必要があります。
2、de_novo_wf Infer de novo tree and decorate with GTDB taxonomy (identify -> align -> infer -> root -> decorate) [In Development]
de novoワークフローでは、ユーザー提供およびGTDB-Tkのファレンスゲノムすべてを含む新しい細菌および古細菌のツリーを推測する。 taxonomy分類ではclassify_wf が推奨される。このワークフローは、de novoでdomain-specificなツリーが必要な場合にのみ推奨される。 このワークフローは、同定、アラインメント、推論、ルート、およびdecorate(まだ実装されていない)で構成される。 同定とアラインメントの手順は、分類ワークフローの場合と同じである。推論ステップでは、FastTreeとWAG + GAMMAモデルを使用して、独立したde novoのバクテリアツリーと古細菌ツリーを計算する。 これらのツリーは、ユーザーが指定したoutgroupを使用してルート化し、GTDB taxonomyでdecorateできる。
gtdbtk de_novo_wf --genome_dir <my_genomes> --<marker_set> --outgroup_taxon <outgroup> --out_dir <output_dir>
コメント
メタゲノムのbinned.fastaファイル群をクエリにして分類を行なってみた。かなり多くの配列が既存の種と同種としてアサインされた。TSVを開き、例えばbinned.fastaの1つchromosome48という配列を見ると、fastaniのアサインがGCF_000025125.1となっている。

出力中のnewlickの系統樹ファイルを開き、このブランチを確認した。phylogenic tree viewerとして、大きなデータセットにも対応したFigtree(Github)を使用した。

確かに同じ位置に存在している。Figtreeでもこのサイズは重いので、目的のゲノムを含むブランチをマウスで囲んで選択し、command + Cでコピーする。

同じブランチの部分がまとめて選択状態になる(一番ルートに近い枝部分を選択すること)。
コピーしたらエディタに貼ってtest.treeとして保存する。

もう一度 、お気に入りのphylogenic tree viewerで開く。
開いたところ。ずいぶん見やすくなった。

GTDB HPについて

Browsesから分類結果を確認できる。
https://gtdb.ecogenomic.org/tree

ブロックをクリックすると、各菌の分類結果のページにジャンプする。

追記
2019年にペーパーになったAnnotreeも有用です。AnnotreeはGTDBの分類を使用したwebの系統樹ビューアです。ビューアですが、アノテーションを行なってタンパク質情報を紐づけているので、GTDBのtaxonomyを調べたい時だけでなく、タンパク質情報から検索する事もできます。素晴らしいツールです。
追記
共通して見出される120の遺伝子(アーキアは122遺伝子)については"Recovery of nearly 8,000 metagenome-assembled genomes substantially expands the tree of life, pubmed"のペーパーで探索されたものが使われている。全リストはこの論文の「supplementary document のtable.S6とS.7を参照(リンク)。
引用
A complete domain-to-species taxonomy for Bacteria and Archaea
Donovan H. Parks, Maria Chuvochina, Pierre-Alain Chaumeil, Christian Rinke, Aaron J. Mussig & Philip Hugenholtz
Nature Biotechnology volume 38, pages1079–1086(2020)
GTDB-Tk: a toolkit to classify genomes with the Genome Taxonomy Database
Pierre-Alain Chaumeil, Aaron J Mussig, Philip Hugenholtz, Donovan H Parks
Bioinformatics, Volume 36, Issue 6, 15 March 2020, Pages 1925–1927
A proposal for a standardized bacterial taxonomy based on genome phylogeny
Donovan H. Parks, Maria Chuvochina, David W. Waite, Christian Rinke, Adam
Skarshewski, Pierre-Alain Chaumeil, Philip Hugenholtz
bioRxiv preprint first posted online Jan. 30, 2018
A standardized bacterial taxonomy based on genome phylogeny substantially revises the tree of life
Donovan H Parks, Maria Chuvochina, David W Waite, Christian Rinke, Adam Skarshewski, Pierre-Alain Chaumeil & Philip Hugenholtz
Nature Biotechnology volume 36, pages 996–1004 (2018)
Recovery of nearly 8,000 metagenome-assembled genomes substantially expands the tree of life
Parks DH, Rinke C, Chuvochina M, Chaumeil PA, Woodcroft BJ, Evans PN, Hugenholtz P, Tyson GW
Nat Microbiol. 2017 Nov;2(11):1533-1542
2021 9/28
GTDB: an ongoing census of bacterial and archaeal diversity through a phylogenetically consistent, rank normalized and complete genome-based taxonomy
Donovan H Parks, Maria Chuvochina, Christian Rinke, Aaron J Mussig, Pierre-Alain Chaumeil, Philip Hugenholtz
Nucleic Acids Res. 2021 Sep 14;gkab776.
Nature Biotechnology volume 38, pages1079–1086(2020)
GTDBにおいて種の代表ゲノムをどのように決定したか説明した論文。ここが大変な部分だが、簡潔にいうとタイプストレインを中心にANIとAF閾値内で1つずつ選ばれている。ANI閾値内で有効なタイプストレインが存在しない場合、ゲノムデータのクオリティの高さで代表が選ばれている。
Bioinformatics, Volume 36, Issue 6, 15 March 2020, Pages 1925–1927
新しいゲノムをGTDBリソースに基づいて分類するためのツールの紹介。
Nature Biotechnology volume 36, pages 996–1004 (2018)
GTDBデータベースの紹介。
2023/08/10
Proposal of names for 329 higher rank taxa defined in the Genome Taxonomy Database under two prokaryotic codes
Maria Chuvochina, Aaron J Mussig, Pierre-Alain Chaumeil, Adam Skarshewski, Christian Rinke, Donovan H Parks, Philip Hugenholtz
FEMS Microbiology Letters, Volume 370, 2023
"2017年11月にGTDB-細菌および古細菌ゲノムの分類を利用できるウェブベースのリソースを提供開始した。GTDBの主な目的は、体系的なゲノムベースのアプローチを用いて、培養および未培養の分類群の完全な分類法(種からドメインまで)を提供することである。(中略)このような新しい名前は、国際原核生物命名規約(ICNP)に基づく有効な出版に必要な分類学的記述を伴わずに、GTDBウェブサイトを通じてのみ紹介されていた。その後、第三者がGTDBのラテン名を暫定的に公表することで混乱が生じたため、この方法を廃止した(Parks et al.2022)(Sanford et al.2021)。(中略)ここでは、GTDB分類学で現在も使用されている属以上のほとんどの暫定的なGTDBラテン語名について、有効なpublicationを準備した。場合によっては、SeqCode下で、ゲノム配列をタイプ材料として提案する必要があった。また、GTDB分類学では、GTDB分類学で現在も使用されている属名を提案する場合もあり、その場合は、SeqCode(下記参照)の下で、ゲノム配列をタイプ材料として提案する。"
関連
参考ページ
*1
FastaniはANI近似値を出せるが、ばらつきもある(paper)。
*2
muliti-fastaにまとめてしまうと正しく動作しない。ゲノムごとに個別のFASTAファイルに分割して保存する。
追記
- GTDB-Tk classify
https://kbase.us/applist/apps/kb_gtdbtk/run_kb_gtdbtk
New Narrativeを選択

ウィンドウを立ち上げる。Add data

Add Dataからゲノムをアップロードして使う。ウィンドウ内にファイルをドラッグアンドドロップして読み込み後、ロードボタンを押すと下のリストにファイルが表示される。それからファイルの種類をAssemblyにして、右隣の上矢印ボタンを押す。

import FASTAアプリが起動し、ファイルがサーバに読み込まれる。

読み込まれると左上のMyDataにファイルが表示され、解析に使用できるようになる。

Tree of Life (TOL)
均等にサンプルされた10,575個の細菌および古細菌ゲノムの参照系統を、複数の戦略を用いて、381個のマーカーの包括的なセットに基づいて構築した。これらは、2017年3月7日時点でNCBI GenBankとRefSeq24から利用可能な86,200の非冗長ゲノムすべてからサンプリングされ、対象となる生物多様性を最大化する統計的アプローチを用いている。