2021 5/16 リンク追加
2021 5/19 インストール手順とコマンドを修正
2022/10/23 追記
全ての微生物集団の配列が同時にサンプリングされるため、メタゲノム試料の個々のゲノムの回収には困難を伴う。しかし、自然生態系および人工生態系における未耕地微生物の機能的可能性を理解することは重要な手順である。ビニングとは、アセンブリされていないリードまたはアセンブリされたコンティグを、1つのビンが1つのゲノムを表す個別のクラスターに分類するプロセスである。主なビニング手法には、ヌクレオチド頻度によるクラスタリング(Dick et al、2009; Iverson et al、2009; Mackelprang et al、2011; Wang et al、2012; Wrighton et al、2012)、まシーケンスカバレッジ(Albertsen et al、2013; Sharon et al、2013; Wu and Ye、2011)がある。ビニングプロセスの自動化は、メタゲノムから微生物ゲノムを回収するためのハイスループットな方法を提供する。著者らは以前に、メタゲノムデータセットからアセンブリされたゲノム配列を自動分類するビニングアルゴリズムMaxBin(Wu et al、2014)を開発した。テトラヌクレオチド頻度と配列カバレッジの両方に基づいて、MaxBinは自動的にターゲットメタゲノムからビン番号を推定し、その配列をゲノムビンに分類し、メタゲノム内のビニングされたゲノムのカバレッジレベルを測定する。MaxBin(Wu et al、2014)を使用して堆肥からエンリッチされた2つのセルロース分解性細菌コンソーシアムから19および26のドラフトゲノムが回収され、未培養微生物のゲノムを回復する有用性が実証された。
複数のサンプルからゲノムを回収すると、ビニングアルゴリズムのパフォーマンスが向上する可能性がある。ゲノムをビンに入れるために、 2つのメタゲノミックサンプルからのコンティグのカバレッジ情報を2次元マップ上にプロットし(Albertsen et al、2013)(紹介)、カバレッジの違いを利用してビニングする手法が開発された。複数のサンプルを使用するビニングツールも、2つ以上のサンプルを共にアセンブリしてビニングする時に優れたパフォーマンスを示した(Alneberg et al、2014; Imelfort et al、2014; Kang et al、2015)。
本論文では、複数のメタゲノムサンプルの共同アセンブリからゲノムを回収するMaxBinアルゴリズムの次世代MaxBin 2.0について説明する。複数のメタゲノミックデータセットにわたるコンティグカバレッジレベルを利用することにより、MaxBin 2.0は個々のメタゲノムサンプルをビニングするよりも優れたビニング結果を実現する。複数のメタゲノミックデータセットを使用する他のビニングアルゴリズムと比較して、MaxBin 2.0は、シミュレートされたメタゲノムからゲノムを回復する際に非常に正確だった。ゲノムビンのカバレッジレベルを測定するMaxBin2.0の能力はまた、複数のサンプルにわたるゲノム - 微生物コミュニティ組成の比較を可能にする。
Methods
MaxBin 2.0は、EMアルゴリズム(wiki)を使用して、メタゲノムからドラフトゲノムを復元する。簡潔に述べると、複数のメタゲノムデータセットのシーケンシングリードを共にアセンブリした後、MaxBin 2.0は、関連するすべてのメタゲノムのコンティグのカバレッジとテトラヌクレオチド頻度を測定し、コンティグを個々のビンに分類する。ビニングプロセスの前に未知であった全てのゲノムビンの存在量は、EMアルゴリズムによって推定される。論文の以下の説明は、複数のメタゲノムデータセットのMaxBinアルゴリズムへの組み込みを示している。
Documnet
https://sourceforge.net/projects/maxbin2/files/

The general workflow of MaxBin. MaxBinのペーパーより転載。
インストール
macではFragGeneScanのinputでエラーがでる。ubuntu16.04のdocker イメージをビルドし、コンテナ上でテストした。
依存
- g++ (c++11 or higher), make
- Perl5 with CPAN, LWP::Simple, and FindBin modules (./autobuild_auxiliary will use CPAN to check and install the other two Perl modules)
- (for automatic downloading and installing auxiliary software) curl, unzip, which
- Bowtie2 (tested version: bowtie2-2.2.3)
- FragGeneScan (tested version: FragGeneScan 1.30)
- Hmmer3 (tested version: HMMER 3.1b1 64 bit)
- IDBA-UD (tested version: IDBA-UD 1.1.1)
本体 SourceForge
#bioconda (link)
mamba create -n maxbin2 -y
conda activate maxbin2
mamba install -c bioconda maxbin2 -y
> run_MaxBin.pl
$ run_MaxBin.pl
MaxBin 2.2.4
No Contig file. Please specify contig file by -contig
MaxBin - a metagenomics binning software.
Usage:
run_MaxBin.pl
-contig (contig file)
-out (output file)
(Input reads and abundance information)
[-reads (reads file) -reads2 (readsfile) -reads3 (readsfile) -reads4 ... ]
[-abund (abundance file) -abund2 (abundfile) -abund3 (abundfile) -abund4 ... ]
(You can also input lists consisting of reads and abundance files)
[-reads_list (list of reads files)]
[-abund_list (list of abundance files)]
(Other parameters)
[-min_contig_length (minimum contig length. Default 1000)]
[-max_iteration (maximum Expectation-Maximization algorithm iteration number. Default 50)]
[-thread (thread num; default 1)]
[-prob_threshold (probability threshold for EM final classification. Default 0.9)]
[-plotmarker]
[-markerset (marker gene sets, 107 (default) or 40. See README for more information.)]
(for debug purpose)
[-version] [-v] (print version number)
[-verbose]
[-preserve_intermediate]
Please specify either -reads or -abund information.
You can input multiple reads and/or abundance files at the same time.
Please read README file for more details.
kamisakBook
ラン
パターン1
ショートリードを指定してランする。fastqはあらかじめcatで結合しておく。Maxbinの内部でbowtieが呼び出され、biningに必要なcoverage情報が計算される。gzip圧縮したfastqにも対応している。
cat sample1_pair1.fq sample1_pair2.fq > sample1_merged.fq
run_MaxBin.pl -contig metagenome_assembly.fa -reads sample1_merged.fq -out output -thread 20
#複数fastqある場合は複数サンプルのカバレッジ情報を使えるのでよりパワフル(ここでは3つ)
cat sample1_R1.fq sample1_R2.fq > sample1_merged.fq
cat sample1_R2.fq sample2_R2.fq > sample2_merged.fq
cat sample3_R2.fq sample3_R2.fq > sample3_merged.fq
run_MaxBin.pl -contig metagenome_assembly.fa -reads sample1_merged.fq -reads2 sample2_merged.fq -reads3 sample3_merged.fq -out output -thread 40
ジョブが終わると、binningされたFASTAファイル、abundanceファイルなどが出力される。目視ではわからない程度の非常に軽度のコンタミネーションがあるバクテリアデータのアセンブリに適用すると、2つのFASTAが出力された。

maxbin2.summary
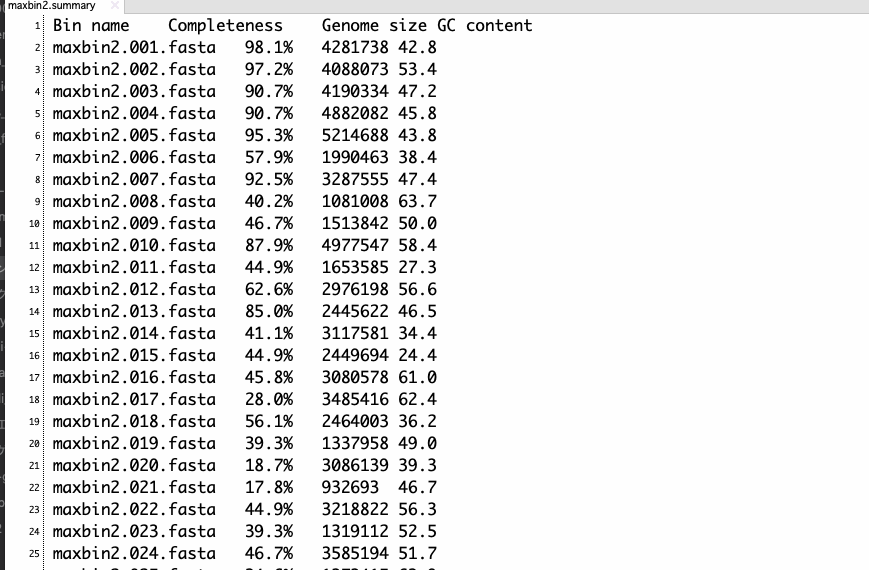
パターン2
bowiteを使わず、BBtoolsでabundanceを別途計算する。それからabundance情報ファイルを指定してランする。
bbmap.sh ref=contigs.fa nodisk in1=pair1.fq in2=pair2.fq covstats=sample.stats
cut -f 1,2 sample.stats > myabund
run_MaxBin.pl -contig contigs.fa -abund myabund -out output -thread 20
docker
ここではnanozooのイメージ(tag)を使う
mkdir maxbin2
docker run -iv $PWD:/data -w /data --rm nanozoo/maxbin2:latest run_MaxBin.pl -contig in.fasta -reads merged.fq.gz -thread 20 -out maxbin2/out_prefix
2021 5/16
Recovering Individual Genomes from Metagenomes Using MaxBin 2.0
https://currentprotocols.onlinelibrary.wiley.com/doi/abs/10.1002/cpz1.128
引用
MaxBin 2.0: an automated binning algorithm to recover genomes from multiple metagenomic datasets
Wu YW, Simmons BA, Singer SW
Bioinformatics. 2016 Feb 15;32(4):605-7.
MaxBin: an automated binning method to recover individual genomes from metagenomes using an expectation-maximization algorithm
Wu YW, Tang YH, Tringe SG, Simmons BA, Singer SW
Microbiome. 2014 Aug 1;2:26.
関連